-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
Autophagy Activators/Inhibitors
- Featured Inhibitors
- Inhibitors (68)
- Modulators (16)
- Chemicals (2)
- Agonists (3)
- Activators (209)
- Antagonist (1)
| Cat.No. | Product Name | Information | Product Use Citations | Product Validations |
|---|---|---|---|---|
| S4430 | HCQ (Hydroxychloroquine) Sulfate | Hydroxychloroquine (HCQ) Sulfate is an antimalarial agent used for the treatment of systemic lupus erythematosus, rheumatoid arthritis and other autoimmune, inflammatory and dermatologic conditions. Also acts as an inhibitor of autophagy and toll-like receptor (TLR) 7/9. |
|
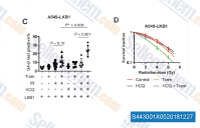
|
| S1396 | Resveratrol (trans-Resveratrol) | Resveratrol has a wide spectrum of targets including cyclooxygenases(i.e. COX, IC50=1.1 μM), lipooxygenases(LOX, IC50=2.7 μM), kinases, sirtuins and other proteins. It has anti-cancer, anti-inflammatory, blood-sugar-lowering and other beneficial cardiovascular effects. Resveratrol induces mitophagy/autophagy and autophagy-dependent apoptosis. |
|
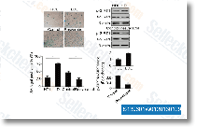
|
| S1049 | Y-27632 Dihydrochloride | Y-27632 2HCl is a selective ROCK1 and ROCK2 inhibitor with a Ki of 140 nM and 300nM in a cell-free assay, exhibits >200-fold selectivity over other kinases, including PKC, cAMP-dependent protein kinase, MLCK and PAK. |
|

|
| S1263 | CHIR-99021 (Laduviglusib) | Laduviglusib (CHIR-99021, CT99021) is a GSK-3α and GSK-3β inhibitor with IC50 values of 10 nM and 6.7 nM, respectively. It does not exhibit cross-reactivity against cyclin-dependent kinases (CDKs) and shows a 350-fold selectivity toward GSK-3β compared to CDKs. This compound functions as a Wnt/β-catenin activator and induces autophagy. |
|
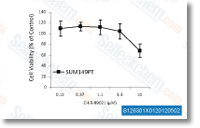
|
| S1060 | AZD2281 (Olaparib) | Olaparib (AZD2281, KU0059436) is a selective inhibitor of PARP1/2 with IC50 of 5 nM/1 nM in cell-free assays, 300-times less effective against tankyrase-1. Olaparib induces significant autophagy that is associated with mitophagy in cells with BRCA mutations. |
|
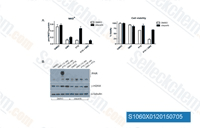
|
| S8048 | ABT-199 (Venetoclax) | Venetoclax (ABT-199, GDC-0199) is a Bcl-2-selective inhibitor with Ki of <0.01 nM in cell-free assays, >4800-fold more selective versus Bcl-xL and Bcl-w, and no activity to Mcl-1. Venetoclax is reported to induce cell growth suppression, apoptosis, cell cycle arrest, and autophagy in triple negative breast cancer MDA-MB-231 cells. Phase 3. |
|
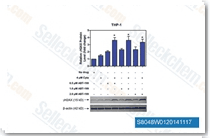
|
| S7655 | Telaglenastat (CB-839) | Telaglenastat (CB-839) is a potent, selective, and orally bioavailable glutaminase inhibitor with IC50 of 24 nM for recombinant human GAC. It induces autophagy and has antitumor activity. Phase 1. |
|
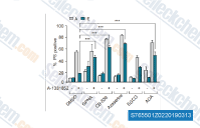
|
| S1378 | Ruxolitinib (INCB18424) | Ruxolitinib (INCB18424) is the first potent, selective, JAK1/2 inhibitor to enter the clinic with IC50 of 3.3 nM/2.8 nM in cell-free assays, >130-fold selectivity for JAK1/2 versus JAK3. This compound kills tumor cells through toxic mitophagy. It induces autophagy and enhances apoptosis. |
|
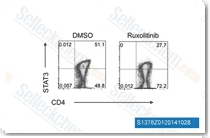
|
| S1039 | Rapamycin (Sirolimus) | Rapamycin is a specific mTOR inhibitor with IC50 of ~0.1 nM in HEK293 cells. This compound binds to FKBP12 and specifically acts as an allosteric inhibitor of mTORC1. It is an autophagy activator and an immunosuppressant. |
|
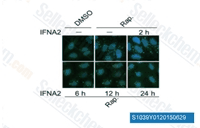
|
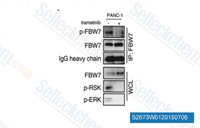
| S2673 | Trametinib (GSK1120212) | Trametinib (GSK1120212, JTP-74057) is a highly specific and potent MEK1/2 inhibitor with IC50 of 0.92 nM/1.8 nM in cell-free assays, and it does not inhibit the kinase activities of c-Raf, B-Raf, ERK1/2. This compound activates autophagy and induces apoptosis. |
|
|
Signaling Pathway Map
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.