-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
research use only
Idarubicin HCl Topoisomerase inhibitor
Cat.No.S1228
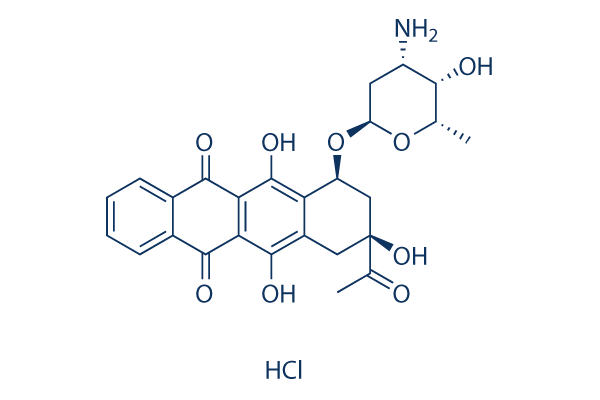
Chemical Structure
Molecular Weight: 533.95
Quality Control
| Related Targets | HDAC PARP ATM/ATR DNA-PK WRN DNA/RNA Synthesis PPAR Sirtuin Casein Kinase eIF |
|---|---|
| Other Topoisomerase Inhibitors | Camptothecin (CPT) Betulinic acid (S)-10-Hydroxycamptothecin Beta-Lapachone Ellagic acid Amonafide Voreloxin (SNS-595) hydrochloride Hydroxy Camptothecine Cu(II)-Elesclomol Genz-644282 |
Cell Culture, Treatment & Working Concentration
| Cell Lines | Assay Type | Concentration | Incubation Time | Formulation | Activity Description | PMID |
|---|---|---|---|---|---|---|
| K562 erythroleukemic cells | Cytotoxic assay | Cytotoxic activity against K562 erythroleukemic cells, IC50=0.002 μM | 10425093 | |||
| K562 cells | Cytotoxic assay | 5 days | Cytotoxicity against human K562 cells after 5 days by XTT assay, IC50=0.012 μM | 18076140 | ||
| mouse DA-3 cells | Cytotoxic assay | 24 h | Cytotoxicity against mouse DA-3 cells after 24 hrs by XTT assay, IC50=2.3 μM | 24900668 | ||
| human ES2 cells | Cytotoxic assay | 24 h | Cytotoxicity against human ES2 cells after 24 hrs by XTT assay, IC50=3.2 μM | 24900668 | ||
| human K562 cells | Proliferation assay | 72 h | Antiproliferative activity against human K562 cells after 72 hrs, GI50=3.3 μM | 25420175 | ||
| human SKOV3 cells | Cytotoxic assay | 24 h | Cytotoxicity against human SKOV3 cells after 24 hrs by XTT assay, IC50=4.5 μM | 24900668 | ||
| HepG2 cells | Function assay | Inhibition of liver stage Plasmodium berghei infection in HepG2 cells, IC50=6.62 μM | 22586124 | |||
| human MCF7 cells | Cytotoxic assay | 24 h | Cytotoxicity against human MCF7 cells after 24 hrs by XTT assay, IC50=7.3 μM | 24900668 | ||
| mouse DA-3 cells | Cytotoxic assay | 20 μM | Cytotoxicity against mouse DA-3 cells assessed as reduction in cell viability at 20 uM by XTT assay | 24900668 | ||
| neural precursor cells | Function assay | Inhibition of neurosphere proliferation of mouse neural precursor cells by MTT assay | 17417631 | |||
| VERO-E6 | Function assay | 48 hrs | Toxicity CC50 against VERO-E6 cells determined at 48 hours by high content imaging (same conditions as 2_LEY without exposure to 0.01 MOI SARS CoV-2 virus), CC50=0.2μM | ChEMBL | ||
| Click to View More Cell Line Experimental Data | ||||||
Solubility
|
In vitro |
DMSO
: 100 mg/mL
(187.28 mM)
Water : 8 mg/mL Ethanol : Insoluble |
Molarity Calculator
|
In vivo |
|||||
In vivo Formulation Calculator (Clear solution)
Step 1: Enter information below (Recommended: An additional animal making an allowance for loss during the experiment)
Step 2: Enter the in vivo formulation (This is only the calculator, not formulation. Please contact us first if there is no in vivo formulation at the solubility Section.)
Calculation results:
Working concentration: mg/ml;
Method for preparing DMSO master liquid: mg drug pre-dissolved in μL DMSO ( Master liquid concentration mg/mL, Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug. )
Method for preparing in vivo formulation: Take μL DMSO master liquid, next addμL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O, mix and clarify.
Method for preparing in vivo formulation: Take μL DMSO master liquid, next add μL Corn oil, mix and clarify.
Note: 1. Please make sure the liquid is clear before adding the next solvent.
2. Be sure to add the solvent(s) in order. You must ensure that the solution obtained, in the previous addition, is a clear solution before proceeding to add the next solvent. Physical methods such
as vortex, ultrasound or hot water bath can be used to aid dissolving.
Chemical Information, Storage & Stability
| Molecular Weight | 533.95 | Formula | C26H27NO9.HCl |
Storage (From the date of receipt) | |
|---|---|---|---|---|---|
| CAS No. | 57852-57-0 | Download SDF | Storage of Stock Solutions |
|
|
| Synonyms | 4-demethoxydaunorubicin (NSC256439, 4-DMDR) HCl | Smiles | CC1C(C(CC(O1)OC2CC(CC3=C2C(=C4C(=C3O)C(=O)C5=CC=CC=C5C4=O)O)(C(=O)C)O)N)O.Cl | ||
Mechanism of Action
| Features |
Idarubicin is a substrate for CYP450 2D6 and 2C9.
|
|---|---|
| Targets/IC50/Ki |
Topo II (MCF-7 cells)
(Cell-free assay) 3.3 ng/mL
Multicellular spheroids
(Cell-free assay) 7.9 ng/mL
|
| In vitro |
Idarubicin has significant cytotoxic activity against multicellular spheroids, comparable to the antiproliferative effects on monolayer cells. Idarubicin inhibits CYP450 2D6. Idarubicin is about 57.5-fold and 25-fold more active than doxorubicin and epirubicin, respectively. Idarubicin is able to overcome P-glycoprotein-mediated multidrug resistance. Idarubicin inhibits PMN superoxide radical formation. Idarubicin could be coupled to the monoclonal antibodies (anti-Ly-2.1, anti-L3T4, or anti-Thy-1) with retention of protein solubility and antibody activity. Idarubicin inhibits the proliferation of NALM-6 cells with an IC50 of 12 nM. |
| Kinase Assay |
CYP450 metabolism experiments
|
|
Evaluation of Idarubicin metabolism by the CYP450 isoenzymes 3A4, 2D6, 2C8, 2C9, and 1A2 is completed using isolated human CYP450 proteins for each isoform. The high throughput P450 inhibition testing method is utilized for these evaluations. The metabolism experiments are designed to investigate the following properties of each drug: (1) if Idarubicin is a substrate of the CYP450 3A4, 2C8, 2C9, 1A2 or 2D6 isoenzymes; (2) if metabolism is affected by known inhibitors of each isoenzyme; (3) if Idarubicin is inhibitors of CYP450 isoenzymes; and (4) if caspofungin or itraconazole inhibit the CYP450 metabolism of Idarubicin. Dibenzylfluorescein (DBF) (CYP3A4, CYP2C8, CYP2C9), 3-cyano-7-ethoxycoumarin (Cyp1A2), and 7-methoxy-4-(aminomethyl)-coumarin (MAMC) (CYP2D6) are the known substrates utilized as controls to confirm the respective isoenzyme activity and evaluate the effects of Idarubicin on the isoenzyme activity. In addition, ketoconazole, quercetin, suflaphenazole, furafylline, and quinidine are utilized as control CYP450 inhibitors for 3A4, 2C8, 2C9, 1A2 or 2D6 isoenzymes, respectively. The substrate, inhibitor plus Idarubicin as indicated are added to each protein sample are incubated for 20 minutes- 60 minutes, as recommend by manufacturer, at 37oC. Reactions are stopped with an organic solvent solution and then samples are analyzed by fluorescence plate reader as appropriate. For each experiment, control samples with a known amount of substrate and synthesized metabolite, in the absence of the isoenzyme, are prepared for qualitative comparisons. All experiments are performed in triplicate.
|
|
| In vivo |
Reduction of Idarubicin is dependent upon ketone reductases, and proceeds more stereoselectively than that of most ketones giving rise to the (13S)-epimer almost exclusively. The high stereospecificity in Idarubicin reduction might result from chiral induction due to the presence of asymmetric centres near to the carbonyl group in Idarubicin. |
References |
|
Clinical Trial Information
(data from https://clinicaltrials.gov, updated on 2024-05-22)
| NCT Number | Recruitment | Conditions | Sponsor/Collaborators | Start Date | Phases |
|---|---|---|---|---|---|
| NCT05697510 | Recruiting | Acute Myeloid Leukemia (AML) |
Nantes University Hospital |
March 16 2023 | Phase 1 |
| NCT02652871 | Completed | Leukemia |
M.D. Anderson Cancer Center|Eli Lilly and Company|High Impact Clinical Research Support Program |
May 9 2016 | Phase 1 |
Tech Support
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
Signaling Pathway Map
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.