-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
research use only
PF-02341066 (Crizotinib) ALK/c-Met/ROS1 Inhibitor
Cat.No.S1068
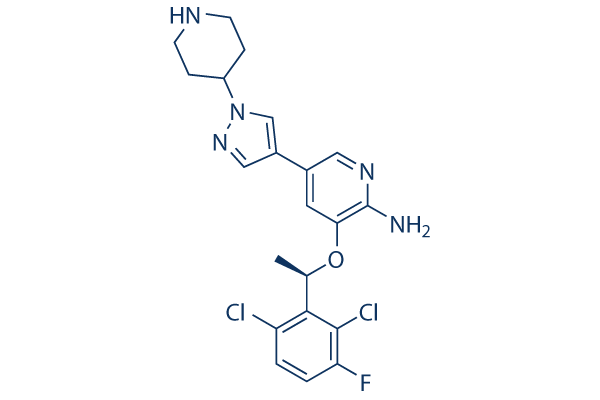
Chemical Structure
Molecular Weight: 450.34
Quality Control
| Related Targets | EGFR VEGFR PDGFR FGFR Src MEK CSF-1R FLT3 HER2 c-Kit |
|---|---|
| Other c-Met Inhibitors | Tepotinib (EMD-1214063) Dihexa SGX-523 PHA-665752 Foretinib (GSK1363089, XL880) SU11274 BMS-777607 JNJ-38877605 Tivantinib PF-04217903 |
Cell Culture, Treatment & Working Concentration
| Cell Lines | Assay Type | Concentration | Incubation Time | Formulation | Activity Description | PMID |
|---|---|---|---|---|---|---|
| SU-DHL1 | Cytotoxic Assay | Cytotoxicity against human SU-DHL1 cells expressing ALK coexpressing NPM with IC50 of 0.01 μM | 21572589 | |||
| BAF3 | Cytotoxic Assay | 48 h | DMSO | Cytotoxicity against mouse BAF3 cells expressing ALK F1174L mutant coexpressing EML4 with IC50 of 0.62 μM | 21572589 | |
| BAF3 | Cytotoxic Assay | 48 h | DMSO | Cytotoxicity against mouse BAF3 cells expressing ALK L1196M mutant coexpressing EML4 with IC50 of 2.2 μM | 21572589 | |
| BAF3 | Cytotoxic Assay | 48 h | DMSO | Cytotoxicity against mouse BAF3 cells expressing EML4-ALK with IC50 of 0.28 μM | 21572589 | |
| Kelly | Cytotoxic Assay | DMSO | Cytotoxicity against human Kelly cells expressing ALK F1174L mutant with IC50 of 0.42 μM | 21572589 | ||
| SH-SY5Y | Cytotoxic Assay | DMSO | Cytotoxicity against human SH-SY5Y cells expressing ALK F1174L mutant with IC50 of 0.53 μM | 21572589 | ||
| SMS-KCN | Cytotoxic Assay | DMSO | Cytotoxicity against human SMS-KCN cells expressing ALK R1275Q mutant with IC50 of 0.91 μM | 21572589 | ||
| BAF3 | Cytotoxic Assay | 48 h | DMSO | Cytotoxicity against mouse BAF3 cells expressing Tel-ALK with IC50 of 0.19 μM | 21572589 | |
| 3T3 | Function Assay | 1 h | DMSO | Inhibition of RON assessed as growth factor-induced autophosphorylation with IC50 of 0.08 μM | 21812414 | |
| 3T3-E | Function Assay | 1 h | DMSO | Inhibition of TIE2 assessed growth factor-induced autophosphorylation with IC50 of 0.448 μM | 21812414 | |
| A549 | Kinase Assay | 1 h | DMSO | Inhibition of human recombinant c-MET kinase expressed assessed as inhibition of HGF-induced autophosphorylation with IC50 of 0.008 μM | 21812414 | |
| BAF3-BCL | Function Assay | 1 h | DMSO | Inhibition of ABL assessed as growth factor-induced autophosphorylation with IC50 of 1.159 μM | 21812414 | |
| HEK293 | Function Assay | 1 h | DMSO | Inhibition of AXL assessed as growth factor-induced autophosphorylation with IC50 of 0.294 μM | 21812414 | |
| HEK293 | Function Assay | 1 h | DMSO | Inhibition of IR assessed as growth factor-induced autophosphorylation with IC50 of 2.887 μM | 21812414 | |
| Jurkat | Function Assay | 1 h | DMSO | Inhibition of LCK assessed as growth factor-induced autophosphorylation with IC50 of 2.741 μM | 21812414 | |
| KARPAS299 | Kinase Assay | 1 h | DMSO | Inhibition of ALK assessed as growth factor-induced autophosphorylation with IC50 of 0.02 μM | 21812414 | |
| PAE | Function Assay | 1 h | DMSO | Inhibition of TRKB assessed as growth factor-induced autophosphorylation with IC50 of 0.399 μM | 21812414 | |
| PAE | Function Assay | 1 h | DMSO | Inhibition of TRKA assessed as growth factor-induced autophosphorylation with IC50 of 0.58 μM | 21812414 | |
| BAF3 | Function Assay | 2-3 d | DMSO | Inhibition of TEL-fused insulin receptor expressed with IC50 of 1.643 μM | 23742252 | |
| BAF3 | Function Assay | 2-3 d | DMSO | Inhibition of NPM-fused ALK (unknown origin) expressed in mouse BAF3 cells after 2 to 3 days by luciferase reporter gene assay with IC50 of 0.1508 μM | 23742252 | |
| BAF3 | Function Assay | 2-3 d | DMSO | Cytotoxicity against mouse BAF3 cells after 2 to 3 days by luciferase reporter gene assay with IC50 of 3.479 μM | 23742252 | |
| KARPAS299 | Cytotoxic Assay | 2-3 d | DMSO | IC50=0.0642 μM | 23742252 | |
| EBC1 | Growth Inhibition Assay | 72 h | DMSO | IC50=0.023 μM | 23993328 | |
| HCT116 | Growth Inhibition Assay | 72 h | DMSO | IC50=14.82 μM | 23993328 | |
| MCF7 | Growth Inhibition Assay | 72 h | DMSO | IC50=9.58 μM | 23993328 | |
| MDA-MB-231 | Growth Inhibition Assay | 72 h | DMSO | IC50=10.8 μM | 23993328 | |
| MKN45 | Growth Inhibition Assay | 72 h | DMSO | IC50=0.013 μM | 23993328 | |
| NCI-H441 | Growth Inhibition Assay | 72 h | DMSO | IC50=17.25 μM | 23993328 | |
| NCI-H661 | Growth Inhibition Assay | 72 h | DMSO | IC50=11.47 μM | 23993328 | |
| SK-MEL-28 | Growth Inhibition Assay | 72 h | DMSO | IC50=10.97 μM | 23993328 | |
| SKOV3 | Growth Inhibition Assay | 72 h | DMSO | IC50=12.85 μM | 23993328 | |
| SNU5 | Growth Inhibition Assay | 72 h | DMSO | IC50=0.016 μM | 23993328 | |
| Antiproliferative assay | EBC1 | 72 hrs | Antiproliferative activity against human EBC1 cells expressing elevated levels of constitutively active c-Met after 72 hrs by SRB assay, IC50 = 0.053 μM. | 22863529 | ||
| Function assay | KARPAS299 | 72 hrs | Inhibition of ALK-fusion driven cell proliferation in human KARPAS299 cells after 72 hrs by CellTiter Glo assay, IC50 = 0.062 μM. | 24432909 | ||
| Function assay | NIH-3T3 | 1 hr | Inhibition of human wild type EML4-fused ALK expressed in mouse NIH-3T3 cells assessed as phosphorylated ALK level after 1 hr by sandwich ELISA, IC50 = 0.08 μM. | 24432909 | ||
| Function assay | NCI-H3122 | 72 hrs | Inhibition of ALK-fusion driven cell proliferation in human NCI-H3122 cells after 72 hrs by CellTiter Glo assay, IC50 = 0.108 μM. | 24432909 | ||
| Function assay | NCI-H2228 | 72 hrs | Inhibition of ALK-fusion driven cell proliferation in human NCI-H2228 cells after 72 hrs by CellTiter Glo assay, IC50 = 0.118 μM. | 24432909 | ||
| Function assay | NIH-3T3 | 1 hr | Inhibition of human EML4-fused ALK F1174L mutant expressed in mouse NIH-3T3 cells assessed as phospho-ALK level after 1 hr by sandwich ELISA, IC50 = 0.165 μM. | 24432909 | ||
| Function assay | NIH-3T3 | 1 hr | Inhibition of human EML4-fused ALK C1156Y mutant expressed in mouse NIH-3T3 cells assessed as phospho-ALK level after 1 hr by sandwich ELISA, IC50 = 0.478 μM. | 24432909 | ||
| Function assay | NIH-3T3 | 1 hr | Inhibition of human EML4-fused ALK G1269A mutant expressed in mouse NIH-3T3 cells assessed as phospho-ALK level after 1 hr by sandwich ELISA, IC50 = 0.605 μM. | 24432909 | ||
| Function assay | NCI-H3122 | 72 hrs | Inhibition of ALK-fusion driven cell proliferation in human NCI-H3122 cells harboring ALK G1269A mutant after 72 hrs by CellTiter Glo assay, IC50 = 0.623 μM. | 24432909 | ||
| Function assay | NIH-3T3 | 1 hr | Inhibition of human EML4-fused ALK S1206Y mutant expressed in mouse NIH-3T3 cells assessed as phospho-ALK level after 1 hr by sandwich ELISA, IC50 = 0.626 μM. | 24432909 | ||
| Function assay | NCI-H3122 | 72 hrs | Inhibition of ALK-fusion driven cell proliferation in human NCI-H3122 cells harboring ALK L1196M mutant after 72 hrs by CellTiter Glo assay, IC50 = 0.838 μM. | 24432909 | ||
| Function assay | NIH-3T3 | 1 hr | Inhibition of human EML4-fused ALK L1196M mutant expressed in mouse NIH-3T3 cells assessed as phospho-ALK level after 1 hr by sandwich ELISA, IC50 = 0.843 μM. | 24432909 | ||
| Function assay | NIH-3T3 | 1 hr | Inhibition of human EML4-fused ALK L1152R mutant expressed in mouse NIH-3T3 cells assessed as phospho-ALK level after 1 hr by sandwich ELISA, IC50 = 1.026 μM. | 24432909 | ||
| Function assay | NIH-3T3 | 1 hr | Inhibition of human EML4-fused ALK 1151Tins mutant expressed in mouse NIH-3T3 cells assessed as phospho-ALK level after 1 hr by sandwich ELISA, IC50 = 3.039 μM. | 24432909 | ||
| Function assay | BAF3 | 72 hrs | Inhibition of NPM/ALK (unknown origin) transfected in mouse BAF3 cells assessed as cell growth inhibition after 72 hrs by [3H]-thymidine incorporation assay, IC50 = 0.051 μM. | 24468632 | ||
| Function assay | BAF3 | 72 hrs | Inhibition of NPM/ALK (unknown origin) transfected in mouse BAF3 cells assessed as cell growth inhibition after 72 hrs by [3H]-thymidine incorporation assay, IC50 = 0.051 μM. | 24468632 | ||
| Function assay | BAF3 | 72 hrs | Inhibition of NPM/ALK L1196M mutant (unknown origin) transfected in mouse BAF3 cells assessed as cell growth inhibition after 72 hrs by [3H]-thymidine incorporation assay, IC50 = 0.26 μM. | 24468632 | ||
| Cytotoxicity assay | BAF3 | 72 hrs | Cytotoxicity against mouse BAF3 cells assessed as growth inhibition after 72 hrs by [3H]-thymidine incorporation assay, IC50 = 0.98 μM. | 24468632 | ||
| Antiproliferative assay | NIH/3T3 | 72 hrs | Antiproliferative activity against crizotinib-resistant mouse NIH/3T3 cells harboring EML4-ALK variant 1 after 72 hrs by MTT assay, IC50 = 0.0954 μM. | 24785465 | ||
| Antiproliferative assay | SUP-M2 | 72 hrs | Antiproliferative activity against human SUP-M2 cells harboring NPM-ALK after 72 hrs by MTT assay, IC50 = 0.174 μM. | 24785465 | ||
| Antiproliferative assay | NIH/3T3 | 72 hrs | Antiproliferative activity against crizotinib-resistant mouse NIH/3T3 cells harboring EML4-ALK variant 1/L1196M mutant after 72 hrs by MTT assay, IC50 = 0.606 μM. | 24785465 | ||
| Function assay | NIH-3T3 | 1 hr | Inhibition of wild type human EML4-fused ALK expressed in mouse NIH-3T3 cells assessed as phosphorylated ALK level after 1 hr by sandwich ELISA, IC50 = 0.08 μM. | 24819116 | ||
| Function assay | NIH-3T3 | 1 hr | Inhibition of human EML4-fused ALK F1174L mutant expressed in mouse NIH-3T3 cells assessed as phospho-ALK level after 1 hr by sandwich ELISA, IC50 = 0.165 μM. | 24819116 | ||
| Function assay | NIH-3T3 | 1 hr | Inhibition of human EML4-fused ALK C1156Y mutant expressed in mouse NIH-3T3 cells assessed as phospho-ALK level after 1 hr by sandwich ELISA, IC50 = 0.478 μM. | 24819116 | ||
| Function assay | NIH-3T3 | 1 hr | Inhibition of human EML4-fused ALK G1269A mutant expressed in mouse NIH-3T3 cells assessed as phospho-ALK level after 1 hr by sandwich ELISA, IC50 = 0.605 μM. | 24819116 | ||
| Function assay | NIH-3T3 | 1 hr | Inhibition of human EML4-fused ALK S1206Y mutant expressed in mouse NIH-3T3 cells assessed as phospho-ALK level after 1 hr by sandwich ELISA, IC50 = 0.626 μM. | 24819116 | ||
| Function assay | NIH-3T3 | 1 hr | Inhibition of human EML4-fused ALK L1196M mutant expressed in mouse NIH-3T3 cells assessed as phospho-ALK level after 1 hr by sandwich ELISA, IC50 = 0.843 μM. | 24819116 | ||
| Function assay | NIH-3T3 | 1 hr | Inhibition of human EML4-fused ALK L1152R mutant expressed in mouse NIH-3T3 cells assessed as phospho-ALK level after 1 hr by sandwich ELISA, IC50 = 1.026 μM. | 24819116 | ||
| Function assay | NIH-3T3 | 1 hr | Inhibition of human EML4-fused ALK G1202R mutant expressed in mouse NIH-3T3 cells assessed as phospho-ALK level after 1 hr by sandwich ELISA, IC50 = 1.148 μM. | 24819116 | ||
| Function assay | NIH-3T3 | 1 hr | Inhibition of human EML4-fused ALK 1151Tins mutant expressed in mouse NIH-3T3 cells assessed as phospho-ALK level after 1 hr by sandwich ELISA, IC50 = 3.039 μM. | 24819116 | ||
| Function assay | MKN845 | 1 hr | Inhibition of c-Met phosphorylation in human MKN845 cells after 1 hr by western blotting, IC50 = 0.02 μM. | 24900750 | ||
| Function assay | MKN845 | 90 mins | Inhibition of c-Met phosphorylation in human MKN845 cells after 90 mins by Sandwich-ELISA, IC50 = 0.02 μM. | 24900750 | ||
| Function assay | karpas 299 | 90 mins | Inhibition of NPM-fused ALK phosphorylation (unknown origin) expressed in human karpas 299 cells after 90 mins by Sandwich-ELISA, IC50 = 0.11 μM. | 24900750 | ||
| Cytotoxicity assay | NCI-H1993 | 48 hrs | Cytotoxicity against human NCI-H1993 cells after 48 hrs by MTT assay, IC50 = 0.061 μM. | 24900830 | ||
| Cytotoxicity assay | NIH/3T3 | 48 hrs | Cytotoxicity against mouse NIH/3T3 cells after 48 hrs by MTT assay, IC50 = 0.364 μM. | 24900830 | ||
| Cytotoxicity assay | A549 | 48 hrs | Cytotoxicity against human A549 cells after 48 hrs by MTT assay, IC50 = 4.084 μM. | 24900830 | ||
| Cytotoxicity assay | NCI-H1975 | 48 hrs | Cytotoxicity against human NCI-H1975 cells after 48 hrs by MTT assay, IC50 = 7.551 μM. | 24900830 | ||
| Antiproliferative assay | EBC1 | 72 hrs | Antiproliferative activity against cMET-amplified human EBC1 cells after 72 hrs, IC50 = 0.0069 μM. | 24900831 | ||
| Antiproliferative assay | KARPAS299 | 72 hrs | Antiproliferative activity against ALK-dependent human KARPAS299 cells after 72 hrs, IC50 = 0.2 μM. | 24900831 | ||
| Antitumor assay | BAF3 | 50 mg/kg | 2 weeks | Antitumor activity against mouse BAF3 cells expressing EML4-ALK fusion protein allografted in nude mouse assessed as tumor growth inhibition at 50 mg/kg, po qd for 2 weeks relative to vehicle-treated control | 24900831 | |
| Antiproliferative assay | EBC1 | 72 hrs | Antiproliferative activity against human EBC1 cells after 72 hrs, IC50 = 0.021 μM. | 25537530 | ||
| Antiproliferative assay | SNU5 | 72 hrs | Antiproliferative activity against human SNU5 cells after 72 hrs, IC50 = 0.0204 μM. | 26005523 | ||
| Antiproliferative assay | EBC1 | 72 hrs | Antiproliferative activity against human EBC1 cells after 72 hrs, IC50 = 0.0218 μM. | 26005523 | ||
| Antiproliferative assay | MKN45 | 72 hrs | Antiproliferative activity against human MKN45 cells after 72 hrs, IC50 = 0.0381 μM. | 26005523 | ||
| Antiproliferative assay | BAF3/TPR-Met | 72 hrs | Antiproliferative activity against mouse BAF3/TPR-Met cells after 72 hrs, IC50 = 0.1274 μM. | 26005523 | ||
| Antiproliferative assay | NCI-H3122 | 72 hrs | Antiproliferative activity against ALK-dependent human NCI-H3122 cells after 72 hrs, IC50 = 0.2612 μM. | 26476749 | ||
| Antiproliferative assay | NCI-H3122 | 72 hrs | Antiproliferative activity against human NCI-H3122 cells after 72 hrs by CellTiter-Glo Luminescent Cell Viability Assay, EC50 = 0.032 μM. | 26568289 | ||
| Function assay | Ba/F3 | 72 hrs | Inhibition of wild type EML4-ALK (unknown origin) expressed in mouse Ba/F3 cells assessed as cell viability after 72 hrs by MTS assay, IC50 = 0.056 μM. | 26568289 | ||
| Function assay | Ba/F3 | 72 hrs | Inhibition of wild type EML4-ALK (unknown origin) expressed in mouse Ba/F3 cells assessed as cell viability after 72 hrs by MTS assay, IC50 = 0.056 μM. | 26568289 | ||
| Function assay | Ba/F3 | 72 hrs | Inhibition of EML4-ALK S1206Y mutant (unknown origin) expressed in mouse Ba/F3 cells assessed as cell viability after 72 hrs by MTS assay, IC50 = 0.065 μM. | 26568289 | ||
| Function assay | Ba/F3 | 72 hrs | Inhibition of EML4-ALK F1174L mutant (unknown origin) expressed in mouse Ba/F3 cells assessed as cell viability after 72 hrs by MTS assay, IC50 = 0.081 μM. | 26568289 | ||
| Function assay | Ba/F3 | 72 hrs | Inhibition of EML4-ALK C1156Y mutant (unknown origin) expressed in mouse Ba/F3 cells assessed as cell viability after 72 hrs by MTS assay, IC50 = 0.144 μM. | 26568289 | ||
| Antiproliferative assay | SMS-KCNR | 72 hrs | Antiproliferative activity against human SMS-KCNR cells expressing EML4-ALK R1275Q mutant after 72 hrs by CellTiter-Glo Luminescent Cell Viability Assay, EC50 = 0.179 μM. | 26568289 | ||
| Antiproliferative assay | Kelly | 72 hrs | Antiproliferative activity against human Kelly cells expressing EML4-ALK F1174L mutant after 72 hrs by CellTiter-Glo Luminescent Cell Viability Assay, EC50 = 0.211 μM. | 26568289 | ||
| Antiproliferative assay | LAN5 | 72 hrs | Antiproliferative activity against human LAN5 cells expressing EML4-ALK R1275Q mutant after 72 hrs by CellTiter-Glo Luminescent Cell Viability Assay, EC50 = 0.232 μM. | 26568289 | ||
| Antiproliferative assay | DFCI76 | 72 hrs | Antiproliferative activity against human DFCI76 cells expressing EML4-ALK L1152R mutant after 72 hrs by CellTiter-Glo Luminescent Cell Viability Assay, EC50 = 0.233 μM. | 26568289 | ||
| Function assay | Ba/F3 | 72 hrs | Inhibition of EML4-ALK G1202R mutant (unknown origin) expressed in mouse Ba/F3 cells assessed as cell viability after 72 hrs by MTS assay, IC50 = 0.328 μM. | 26568289 | ||
| Antiproliferative assay | SK-N-SH | 72 hrs | Antiproliferative activity against human SK-N-SH cells expressing EML4-ALK F1174L mutant after 72 hrs by CellTiter-Glo Luminescent Cell Viability Assay, EC50 = 0.37 μM. | 26568289 | ||
| Antiproliferative assay | CHLA20 | 72 hrs | Antiproliferative activity against human CHLA20 cells expressing EML4-ALK R1275Q mutant after 72 hrs by CellTiter-Glo Luminescent Cell Viability Assay, EC50 = 0.439 μM. | 26568289 | ||
| Function assay | Ba/F3 | 72 hrs | Inhibition of EML4-ALK G1269A mutant (unknown origin) expressed in mouse Ba/F3 cells assessed as cell viability after 72 hrs by MTS assay, IC50 = 0.512 μM. | 26568289 | ||
| Antiproliferative assay | SH-SY5Y | 72 hrs | Antiproliferative activity against human SH-SY5Y cells expressing EML4-ALK F1174L mutant after 72 hrs by CellTiter-Glo Luminescent Cell Viability Assay, EC50 = 0.523 μM. | 26568289 | ||
| Function assay | Ba/F3 | 72 hrs | Inhibition of EML4-ALK L1196M mutant (unknown origin) expressed in mouse Ba/F3 cells assessed as cell viability after 72 hrs by MTS assay, IC50 = 0.549 μM. | 26568289 | ||
| Function assay | Ba/F3 | 72 hrs | Inhibition of EML4-ALK L1152R mutant (unknown origin) expressed in mouse Ba/F3 cells assessed as cell viability after 72 hrs by MTS assay, IC50 = 0.645 μM. | 26568289 | ||
| Antiproliferative assay | SK-N-BE(2) | 72 hrs | Antiproliferative activity against human SK-N-BE(2) cells expressing wild type EML4-ALK after 72 hrs by CellTiter-Glo Luminescent Cell Viability Assay, EC50 = 0.71 μM. | 26568289 | ||
| Function assay | Ba/F3 | 72 hrs | Inhibition of EML4-ALK 1151Tins mutant (unknown origin) expressed in mouse Ba/F3 cells assessed as cell viability after 72 hrs by MTS assay, IC50 = 0.857 μM. | 26568289 | ||
| Cytotoxicity assay | BA/F3 | 72 hrs | Cytotoxicity against mouse BA/F3 cells assessed as cell viability after 72 hrs by MTS assay, IC50 = 0.927 μM. | 26568289 | ||
| Antiproliferative assay | LAN1 | 72 hrs | Antiproliferative activity against human LAN1 cells expressing EML4-ALK F1174L mutant after 72 hrs by CellTiter-Glo Luminescent Cell Viability Assay, EC50 = 1.346 μM. | 26568289 | ||
| Antiproliferative assay | SK-N-FI | 72 hrs | Antiproliferative activity against human SK-N-FI cells expressing wild type EML4-ALK after 72 hrs by CellTiter-Glo Luminescent Cell Viability Assay, EC50 = 1.469 μM. | 26568289 | ||
| Antiproliferative assay | SK-N-AS | 72 hrs | Antiproliferative activity against human SK-N-AS cells expressing wild type EML4-ALK after 72 hrs by CellTiter-Glo Luminescent Cell Viability Assay, EC50 = 1.473 μM. | 26568289 | ||
| Antiproliferative assay | DFCI114 | 72 hrs | Antiproliferative activity against human DFCI114 cells expressing EML4-ALK G1269A mutant after 72 hrs by CellTiter-Glo Luminescent Cell Viability Assay, EC50 = 1.615 μM. | 26568289 | ||
| Antiproliferative assay | EBC1 | 72 hrs | Antiproliferative activity against human EBC1 cells after 72 hrs by SRB assay, IC50 = 0.019 μM. | 26698536 | ||
| Cytotoxicity assay | EBC1 | 72 hrs | Cytotoxicity against human EBC1 cells assessed as inhibition of cell proliferation after 72 hrs by sulforhodamine B assay, IC50 = 0.044 μM. | 27017548 | ||
| Antiproliferative assay | KARPAS299 | 72 hrs | Antiproliferative activity against human KARPAS299 cells after 72 hrs by SRB/CCK-8 assay, IC50 = 0.103 μM. | 27131066 | ||
| Antiproliferative assay | SUP-M2 | 72 hrs | Antiproliferative activity against human SUP-M2 cells after 72 hrs by SRB/CCK-8 assay, IC50 = 0.112 μM. | 27131066 | ||
| Antiproliferative assay | SU-DHL1 | 72 hrs | Antiproliferative activity against human SU-DHL1 cells after 72 hrs by SRB/CCK-8 assay, IC50 = 0.136 μM. | 27131066 | ||
| Antiproliferative assay | NCI-H3122 | 72 hrs | Antiproliferative activity against human NCI-H3122 cells after 72 hrs by SRB/CCK-8 assay, IC50 = 0.21 μM. | 27131066 | ||
| Function assay | NCI-H3122 | 72 hrs | Inhibition of ALK expressed in human NCI-H3122 cells assessed as cell growth inhibition after 72 hrs by SRB/CCK-8 assay, IC50 = 0.261 μM. | 27131066 | ||
| Antiproliferative assay | NIH/3T3 | 72 hrs | Antiproliferative activity against mouse NIH/3T3 cells expressing wild type EML4-ALK after 72 hrs by SRB/CCK-8 assay, IC50 = 0.283 μM. | 27131066 | ||
| Antiproliferative assay | NIH/3T3 | 72 hrs | Antiproliferative activity against mouse NIH/3T3 cells expressing EML4-ALK L1196 mutant after 72 hrs by SRB/CCK-8 assay, IC50 = 1.16 μM. | 27131066 | ||
| Antiproliferative assay | ALK-positive KARPAS299 | 72 hrs | Antiproliferative activity against human ALK-positive KARPAS299 cells assessed as reduction in cell viability measured after 72 hrs by CellTiter 96 aqueous one solution cell proliferation assay, IC50 = 0.365 μM. | 27144831 | ||
| Antiproliferative assay | ALK-negative U937 | 72 hrs | Antiproliferative activity against human ALK-negative U937 cells assessed as reduction in cell viability measured after 72 hrs by CellTiter 96 aqueous one solution cell proliferation assay, IC50 = 2.286 μM. | 27144831 | ||
| Cytotoxicity assay | HepG2 | 72 hrs | Cytotoxicity against human HepG2 cells assessed as growth inhibition after 72 hrs by MTT assay, IC50 = 3.7 μM. | 27396929 | ||
| Cytotoxicity assay | MIAPaCa2 | 72 hrs | Cytotoxicity against human MIAPaCa2 cells assessed as growth inhibition after 72 hrs by MTT assay, IC50 = 7.16 μM. | 27396929 | ||
| Cytotoxicity assay | HCC827 | 72 hrs | Cytotoxicity against human HCC827 cells assessed as growth inhibition after 72 hrs by MTT assay, IC50 = 7.25 μM. | 27396929 | ||
| Cytotoxicity assay | KARPAS299 | 72 hrs | Cytotoxicity against human KARPAS299 cells harboring NPM-ALK assessed as reduction in cell proliferation after 72 hrs by MTT assay, IC50 = 0.068 μM. | 27474925 | ||
| Cytotoxicity assay | HCC78 | 72 hrs | Cytotoxicity against human HCC78 cells harboring SLC34A2-ROS1 assessed as reduction in cell proliferation after 72 hrs by MTT assay, IC50 = 0.34 μM. | 27474925 | ||
| Antiproliferative assay | SU-DHL1 | 72 hrs | Antiproliferative activity against ALK constitutively activated human SU-DHL1 cells after 72 hrs by SRB or CCK8 assay, IC50 = 0.0923 μM. | 27769623 | ||
| Antiproliferative assay | NCI-H3122 | 72 hrs | Antiproliferative activity against ALK constitutively activated human NCI-H3122 cells after 72 hrs by SRB or CCK8 assay, IC50 = 0.1009 μM. | 27769623 | ||
| Antiproliferative assay | KARPAS299 | 72 hrs | Antiproliferative activity against ALK constitutively activated human KARPAS299 cells after 72 hrs by SRB or CCK8 assay, IC50 = 0.1049 μM. | 27769623 | ||
| Function assay | NIH/3T3 | Inhibition of wild type EML4/ALK (unknown origin) expressed in NIH/3T3 cells, IC50 = 0.08 μM. | 28431340 | |||
| Function assay | NIH/3T3 | Inhibition of wild type EML4/ALK (unknown origin) expressed in NIH/3T3 cells, IC50 = 0.08 μM. | 28431340 | |||
| Function assay | NIH/3T3 | Inhibition of wild type EML4/ALK F1174L mutant (unknown origin) expressed in NIH/3T3 cells, IC50 = 0.165 μM. | 28431340 | |||
| Function assay | NIH/3T3 | Inhibition of wild type EML4/ALK F1174L mutant (unknown origin) expressed in NIH/3T3 cells, IC50 = 0.165 μM. | 28431340 | |||
| Function assay | NIH/3T3 | Inhibition of wild type EML4/ALK C1156Y mutant (unknown origin) expressed in NIH/3T3 cells, IC50 = 0.478 μM. | 28431340 | |||
| Function assay | NIH/3T3 | Inhibition of wild type EML4/ALK C1156Y mutant (unknown origin) expressed in NIH/3T3 cells, IC50 = 0.478 μM. | 28431340 | |||
| Function assay | NIH/3T3 | Inhibition of wild type EML4/ALK G1269A mutant (unknown origin) expressed in NIH/3T3 cells, IC50 = 0.605 μM. | 28431340 | |||
| Function assay | NIH/3T3 | Inhibition of wild type EML4/ALK G1269A mutant (unknown origin) expressed in NIH/3T3 cells, IC50 = 0.605 μM. | 28431340 | |||
| Function assay | NIH/3T3 | Inhibition of wild type EML4/ALK S1206Y mutant (unknown origin) expressed in NIH/3T3 cells, IC50 = 0.626 μM. | 28431340 | |||
| Function assay | NIH/3T3 | Inhibition of wild type EML4/ALK S1206Y mutant (unknown origin) expressed in NIH/3T3 cells, IC50 = 0.626 μM. | 28431340 | |||
| Function assay | NIH/3T3 | Inhibition of wild type EML4/ALK L1196M mutant (unknown origin) expressed in NIH/3T3 cells, IC50 = 0.843 μM. | 28431340 | |||
| Function assay | NIH/3T3 | Inhibition of wild type EML4/ALK L1196M mutant (unknown origin) expressed in NIH/3T3 cells, IC50 = 0.843 μM. | 28431340 | |||
| Function assay | NIH/3T3 | Inhibition of wild type EML4/ALK L1152R mutant (unknown origin) expressed in NIH/3T3 cells, IC50 = 1.026 μM. | 28431340 | |||
| Function assay | NIH/3T3 | Inhibition of wild type EML4/ALK L1152R mutant (unknown origin) expressed in NIH/3T3 cells, IC50 = 1.026 μM. | 28431340 | |||
| Function assay | NIH/3T3 | Inhibition of wild type EML4/ALK G1202R mutant (unknown origin) expressed in NIH/3T3 cells, IC50 = 1.148 μM. | 28431340 | |||
| Function assay | NIH/3T3 | Inhibition of wild type EML4/ALK G1202R mutant (unknown origin) expressed in NIH/3T3 cells, IC50 = 1.148 μM. | 28431340 | |||
| Function assay | NIH/3T3 | Inhibition of wild type EML4/ALK 1151Tins mutant (unknown origin) expressed in NIH/3T3 cells, IC50 = 3.039 μM. | 28431340 | |||
| Function assay | NIH/3T3 | Inhibition of wild type EML4/ALK 1151Tins mutant (unknown origin) expressed in NIH/3T3 cells, IC50 = 3.039 μM. | 28431340 | |||
| Cytotoxicity assay | EBC1 | 72 hrs | Cytotoxicity against human EBC1 cells assessed as decrease in cell viability after 72 hrs by Cell Titer-Glo assay, Activity = 0.013 μM. | 28755635 | ||
| Cytotoxicity assay | MKN45 | 72 hrs | Cytotoxicity against human MKN45 cells assessed as decrease in cell viability after 72 hrs by Cell Titer-Glo assay, Activity = 0.022 μM. | 28755635 | ||
| Cytotoxicity assay | PC3 | 72 hrs | Cytotoxicity against human PC3 cells assessed as decrease in cell viability after 72 hrs by Cell Titer-Glo assay, Activity = 2.244 μM. | 28755635 | ||
| Function assay | BAF3 | 72 hrs | Inhibition of EML4-fused ALK (unknown origin) expressed in mouse BAF3 cells assessed as decrease in cell proliferation after 72 hrs by CellTiter-Glo assay, GI50 = 0.0003 μM. | 28850922 | ||
| Function assay | BAF3 | 72 hrs | Inhibition of NPM-fused ALK (unknown origin) expressed in mouse BAF3 cells assessed as decrease in cell proliferation after 72 hrs by CellTiter-Glo assay, GI50 = 0.0003 μM. | 28850922 | ||
| Function assay | BAF3 | 72 hrs | Inhibition of TEL-fused ALK (unknown origin) expressed in mouse BAF3 cells assessed as decrease in cell proliferation after 72 hrs by CellTiter-Glo assay, GI50 = 0.0003 μM. | 28850922 | ||
| Antiproliferative assay | NCI-H3122 | 72 hrs | Antiproliferative activity against human NCI-H3122 cells harboring EML4-fused ALK varian1 after 72 hrs by CellTiter-Glo assay, GI50 = 0.037 μM. | 28850922 | ||
| Antiproliferative assay | NCI-H2228 | 72 hrs | Antiproliferative activity against human NCI-H2228 cells harboring EML4-fused ALK varian3 after 72 hrs by CellTiter-Glo assay, GI50 = 0.073 μM. | 28850922 | ||
| Function assay | BAF3 | 72 hrs | Inhibition of TEL-fused ALK C1156Y mutant (unknown origin) expressed in mouse BAF3 cells assessed as decrease in cell proliferation after 72 hrs by CellTiter-Glo assay, GI50 = 0.15 μM. | 28850922 | ||
| Function assay | BAF3 | 72 hrs | Inhibition of full length ALK F1174L mutant (unknown origin) expressed in mouse BAF3 cells assessed as decrease in cell proliferation after 72 hrs by CellTiter-Glo assay, GI50 = 0.32 μM. | 28850922 | ||
| Function assay | BAF3 | 72 hrs | Inhibition of TEL-fused ALK G1202R mutant (unknown origin) expressed in mouse BAF3 cells assessed as decrease in cell proliferation after 72 hrs by CellTiter-Glo assay, GI50 = 0.43 μM. | 28850922 | ||
| Antiproliferative assay | CHL | 72 hrs | Antiproliferative activity against CHL cells after 72 hrs by CellTiter-Glo assay, GI50 = 0.45 μM. | 28850922 | ||
| Function assay | BAF3 | 72 hrs | Inhibition of TEL-fused ALK L1196M mutant (unknown origin) expressed in mouse BAF3 cells assessed as decrease in cell proliferation after 72 hrs by CellTiter-Glo assay, GI50 = 0.59 μM. | 28850922 | ||
| Antiproliferative assay | BAF3 | 72 hrs | Antiproliferative activity against mouse BAF3 cells after 72 hrs by CellTiter-Glo assay, GI50 = 1.1 μM. | 28850922 | ||
| Antiproliferative assay | CHO | 72 hrs | Antiproliferative activity against CHO cells after 72 hrs by CellTiter-Glo assay, GI50 = 3.2 μM. | 28850922 | ||
| Antiproliferative assay | NCI-H2228 | 72 hrs | Antiproliferative activity against human NCI-H2228 cells after 72 hrs by MTT assay, IC50 = 2.5 μM. | 29091425 | ||
| Antiproliferative assay | H2228/CR | 72 hrs | Antiproliferative activity against human H2228/CR cells after 72 hrs by MTT assay, IC50 = 10 μM. | 29091425 | ||
| Function assay | H2228 | 0.5 uM | 3 hrs | Inhibition of ALK in human H2228 cells assessed as decrease in AKT phosphorylation at 0.5 uM after 3 hrs by Western blot method | 29091425 | |
| Function assay | Ba/F3 | 0.1 to 1 uM | 72 hrs | Inhibition of human wild type EML4 fused ALK expressed in mouse Ba/F3 cells assessed as decrease in cell proliferation at 0.1 to 1 uM preincubated for 72 hrs followed by methyl-3H-thymidine incorporation measured after 8 hrs by filter scintillation counte | 29091425 | |
| Antiproliferative assay | KARPAS299 | 72 hrs | Antiproliferative activity against human KARPAS299 cells harboring NPM-ALK after 72 hrs by MTT assay, IC50 = 0.087 μM. | 29174809 | ||
| Antiproliferative assay | HCC78 | 72 hrs | Antiproliferative activity against human HCC78 cells harboring SLC34A2-ROS1 after 72 hrs by MTT assay, IC50 = 0.17 μM. | 29174809 | ||
| Antiproliferative assay | NCI-H2228 | 72 hrs | Antiproliferative activity against human NCI-H2228 cells harboring EML4-ALK after 72 hrs by MTT assay, IC50 = 0.24 μM. | 29174809 | ||
| Antiproliferative assay | NCI-H3122 | 48 hrs | Antiproliferative activity against human NCI-H3122 cells after 48 hrs by MTT assay, IC50 = 0.8 μM. | 29174814 | ||
| Antiproliferative assay | HCC78 | 48 hrs | Antiproliferative activity against human HCC78 cells after 48 hrs by MTT assay, IC50 = 2 μM. | 29174814 | ||
| Antiproliferative assay | EBC1 | 72 hrs | Antiproliferative activity against human EBC1 cells after 72 hrs by Alamarblue assay, IC50 = 0.013 μM. | 29202410 | ||
| Antiproliferative assay | MKN45 | 72 hrs | Antiproliferative activity against human MKN45 cells after 72 hrs by Alamarblue assay, IC50 = 0.022 μM. | 29202410 | ||
| Antiproliferative assay | MCF7 | 72 hrs | Antiproliferative activity against human MCF7 cells after 72 hrs by Alamarblue assay relative to control, IC50 = 0.045 μM. | 29202410 | ||
| Antiproliferative assay | A549 | 72 hrs | Antiproliferative activity against human A549 cells after 72 hrs by Alamarblue assay relative to control, IC50 = 0.1343 μM. | 29202410 | ||
| Antiproliferative assay | HCT116 | 72 hrs | Antiproliferative activity against human HCT116 cells after 72 hrs by Alamarblue assay relative to control, IC50 = 0.2536 μM. | 29202410 | ||
| Antiproliferative assay | SGC7901 | 72 hrs | Antiproliferative activity against human SGC7901 cells after 72 hrs by Alamarblue assay relative to control, IC50 = 0.3213 μM. | 29202410 | ||
| Antiproliferative assay | NCI-H460 | 72 hrs | Antiproliferative activity against human NCI-H460 cells after 72 hrs by Alamarblue assay, IC50 = 2.244 μM. | 29202410 | ||
| Antiproliferative assay | COLO205 | 72 hrs | Antiproliferative activity against human COLO205 cells after 72 hrs by Alamarblue assay, IC50 = 2.449 μM. | 29202410 | ||
| Antiproliferative assay | PC3 | 72 hrs | Antiproliferative activity against human PC3 cells after 72 hrs by Alamarblue assay, IC50 = 9.787 μM. | 29202410 | ||
| Antiproliferative assay | BAF3 | 72 hrs | Antiproliferative activity against mouse BAF3 cells harboring CD74-ROS1 after 72 hrs by SRB or CCK8 assay, IC50 = 0.0486 μM. | 29288940 | ||
| Antiproliferative assay | BAF3 | 72 hrs | Antiproliferative activity against mouse BAF3 cells harboring EML4-ALK after 72 hrs by SRB or CCK8 assay, IC50 = 0.0575 μM. | 29288940 | ||
| Antiproliferative assay | SU-DHL1 | 72 hrs | Antiproliferative activity against human SU-DHL1 cells harboring NPM-ALK after 72 hrs by SRB or CCK8 assay, IC50 = 0.155 μM. | 29288940 | ||
| Antiproliferative assay | KARPAS299 | 72 hrs | Antiproliferative activity against human KARPAS299 cells harboring NPM-ALK after 72 hrs by SRB or CCK8 assay, IC50 = 0.176 μM. | 29288940 | ||
| Antiproliferative assay | NCI-H3122 | 72 hrs | Antiproliferative activity against human NCI-H3122 cells after 72 hrs by SRB or CCK8 assay, IC50 = 0.303 μM. | 29288940 | ||
| Antiproliferative assay | BAF3 | 72 hrs | Antiproliferative activity against mouse BAF3 cells harboring EML4-ALK L1196M mutant after 72 hrs by SRB or CCK8 assay, IC50 = 0.34 μM. | 29288940 | ||
| Antiproliferative assay | BAF3 | 72 hrs | Antiproliferative activity against mouse BAF3 cells harboring EML4-ALK G1202R mutant after 72 hrs by SRB or CCK8 assay, IC50 = 0.564 μM. | 29288940 | ||
| Antiproliferative assay | BAF3 | 72 hrs | Antiproliferative activity against mouse BAF3 cells harboring CD74-ROS1 G2032R mutant after 72 hrs by SRB or CCK8 assay, IC50 = 0.594 μM. | 29288940 | ||
| Antiproliferative assay | BAF3 | 72 hrs | Antiproliferative activity against IL3-stimulated mouse BAF3 cells after 72 hrs by SRB or CCK8 assay, IC50 = 0.644 μM. | 29288940 | ||
| Antiproliferative assay | HCC78 | 72 hrs | Antiproliferative activity against human HCC78 cells harboring SLC34A2-ROS1 after 72 hrs by SRB or CCK8 assay, IC50 = 0.889 μM. | 29288940 | ||
| qHTS assay | TC32 | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for TC32 cells | 29435139 | |||
| qHTS assay | A673 | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for A673 cells | 29435139 | |||
| qHTS assay | Saos-2 | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for Saos-2 cells | 29435139 | |||
| qHTS assay | RD | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for RD cells | 29435139 | |||
| qHTS assay | SK-N-SH | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SK-N-SH cells | 29435139 | |||
| qHTS assay | BT-12 | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for BT-12 cells | 29435139 | |||
| qHTS assay | MG 63 (6-TG R) | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for MG 63 (6-TG R) cells | 29435139 | |||
| qHTS assay | NB1643 | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for NB1643 cells | 29435139 | |||
| qHTS assay | OHS-50 | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for OHS-50 cells | 29435139 | |||
| qHTS assay | LAN-5 | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for LAN-5 cells | 29435139 | |||
| qHTS assay | NB-EBc1 | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for NB-EBc1 cells | 29435139 | |||
| qHTS assay | SK-N-SH | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for SK-N-SH cells | 29435139 | |||
| qHTS assay | Rh41 | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for Rh41 cells | 29435139 | |||
| qHTS assay | A673 | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for A673 cells) | 29435139 | |||
| qHTS assay | Rh30 | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for Rh30 cells | 29435139 | |||
| qHTS assay | BT-37 | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for BT-37 cells | 29435139 | |||
| qHTS assay | MG 63 (6-TG R) | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for MG 63 (6-TG R) cells | 29435139 | |||
| qHTS assay | Rh30 | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for Rh30 cells | 29435139 | |||
| qHTS assay | OHS-50 | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for OHS-50 cells | 29435139 | |||
| qHTS assay | SJ-GBM2 | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SJ-GBM2 cells | 29435139 | |||
| qHTS assay | SK-N-MC | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SK-N-MC cells | 29435139 | |||
| qHTS assay | NB-EBc1 | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for NB-EBc1 cells | 29435139 | |||
| qHTS assay | LAN-5 | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for LAN-5 cells | 29435139 | |||
| qHTS assay | Rh18 | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for Rh18 cells | 29435139 | |||
| qHTS assay | NB1643 | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for NB1643 cells | 29435139 | |||
| qHTS assay | SK-N-MC | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for SK-N-MC cells | 29435139 | |||
| qHTS assay | TC32 | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for TC32 cells | 29435139 | |||
| qHTS assay | Rh18 | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for Rh18 cells | 29435139 | |||
| qHTS assay | Saos-2 | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for Saos-2 cells | 29435139 | |||
| Antiproliferative assay | EBC1 | 72 hrs | Antiproliferative activity against human EBC1 cells after 72 hrs by Cell Titer-Glo assay, IC50 = 0.039 μM. | 29602036 | ||
| Function assay | Hs578T | 100 nM | 30 mins | Inhibition of hepsin-mediated conversion of Pro-HGF into active form in human Hs578T cells assessed as decrease in MET phosphorylation at 100 nM preincubated for 30 mins followed by recombinant human pro-HGF addition measured after 30 mins by immunoblot m | 29701962 | |
| Function assay | HCC1937 | 100 nM | 30 mins | Inhibition of hepsin-mediated conversion of Pro-HGF into active form in human HCC1937 cells assessed as decrease in MET phosphorylation at 100 nM preincubated for 30 mins followed by recombinant human pro-HGF addition measured after 30 mins by immunoblot | 29701962 | |
| Antiproliferative assay | NCI-H2228 | 72 hrs | Antiproliferative activity against human NCI-H2228 cells harboring EML4-ALK after 72 hrs by MTT assay, IC50 = 0.087 μM. | 30223120 | ||
| Antiproliferative assay | HCC78 | 72 hrs | Antiproliferative activity against human HCC78 cells harboring SLC34A2-ROS1 after 72 hrs by MTT assay, IC50 = 0.17 μM. | 30223120 | ||
| Antiproliferative assay | KARPAS299 | 72 hrs | Antiproliferative activity against human KARPAS299 cells harboring NPM-ALK after 72 hrs by MTT assay, IC50 = 0.24 μM. | 30223120 | ||
| NB1 | Growth Inhibition Assay | IC50=91.98 nM | SANGER | |||
| NCI-SNU-5 | Growth Inhibition Assay | IC50=105.75 nM | SANGER | |||
| SR | Growth Inhibition Assay | IC50=126.31 nM | SANGER | |||
| SF539 | Growth Inhibition Assay | IC50=204.24 nM | SANGER | |||
| SU-DHL-1 | Growth Inhibition Assay | IC50=336.82 nM | SANGER | |||
| SCC-3 | Growth Inhibition Assay | IC50=356.76 nM | SANGER | |||
| DEL | Growth Inhibition Assay | IC50=369.9 nM | SANGER | |||
| CTV-1 | Growth Inhibition Assay | IC50=596.48 nM | SANGER | |||
| EM-2 | Growth Inhibition Assay | IC50=601.34 nM | SANGER | |||
| MHH-CALL-2 | Growth Inhibition Assay | IC50=682.57 nM | SANGER | |||
| KM12 | Growth Inhibition Assay | IC50=706.9 nM | SANGER | |||
| KINGS-1 | Growth Inhibition Assay | IC50=749.75 nM | SANGER | |||
| MEG-01 | Growth Inhibition Assay | IC50=857.66 nM | SANGER | |||
| BV-173 | Growth Inhibition Assay | IC50=1.05997 μM | SANGER | |||
| LAMA-84 | Growth Inhibition Assay | IC50=1.38282 μM | SANGER | |||
| KARPAS-299 | Growth Inhibition Assay | IC50=1.40861 μM | SANGER | |||
| K-562 | Growth Inhibition Assay | IC50=1.72269 μM | SANGER | |||
| SK-LMS-1 | Growth Inhibition Assay | IC50=1.76867 μM | SANGER | |||
| MOLT-16 | Growth Inhibition Assay | IC50=1.95575 μM | SANGER | |||
| CMK | Growth Inhibition Assay | IC50=1.96159 μM | SANGER | |||
| ST486 | Growth Inhibition Assay | IC50=2.43073 μM | SANGER | |||
| CI-1 | Growth Inhibition Assay | IC50=2.49659 μM | SANGER | |||
| KP-N-RT-BM-1 | Growth Inhibition Assay | IC50=2.70122 μM | SANGER | |||
| ALL-PO | Growth Inhibition Assay | IC50=3.18207 μM | SANGER | |||
| KS-1 | Growth Inhibition Assay | IC50=3.21225 μM | SANGER | |||
| Becker | Growth Inhibition Assay | IC50=4.2393 μM | SANGER | |||
| GDM-1 | Growth Inhibition Assay | IC50=4.24617 μM | SANGER | |||
| BC-1 | Growth Inhibition Assay | IC50=4.49277 μM | SANGER | |||
| NB14 | Growth Inhibition Assay | IC50=4.83524 μM | SANGER | |||
| NOS-1 | Growth Inhibition Assay | IC50=5.33874 μM | SANGER | |||
| MZ1-PC | Growth Inhibition Assay | IC50=5.82151 μM | SANGER | |||
| A498 | Growth Inhibition Assay | IC50=6.08473 μM | SANGER | |||
| EW-16 | Growth Inhibition Assay | IC50=6.37773 μM | SANGER | |||
| NALM-6 | Growth Inhibition Assay | IC50=6.68387 μM | SANGER | |||
| EB-3 | Growth Inhibition Assay | IC50=7.07233 μM | SANGER | |||
| 697 | Growth Inhibition Assay | IC50=9.24329 μM | SANGER | |||
| Ramos-2G6-4C10 | Growth Inhibition Assay | IC50=9.59842 μM | SANGER | |||
| KNS-81-FD | Growth Inhibition Assay | IC50=9.69653 μM | SANGER | |||
| HUTU-80 | Growth Inhibition Assay | IC50=9.74642 μM | SANGER | |||
| LS-411N | Growth Inhibition Assay | IC50=10.0567 μM | SANGER | |||
| RPMI-8402 | Growth Inhibition Assay | IC50=10.116 μM | SANGER | |||
| KU812 | Growth Inhibition Assay | IC50=10.2991 μM | SANGER | |||
| EW-1 | Growth Inhibition Assay | IC50=10.4425 μM | SANGER | |||
| HC-1 | Growth Inhibition Assay | IC50=10.4844 μM | SANGER | |||
| NB69 | Growth Inhibition Assay | IC50=10.5043 μM | SANGER | |||
| MFH-ino | Growth Inhibition Assay | IC50=10.8303 μM | SANGER | |||
| CCRF-CEM | Growth Inhibition Assay | IC50=11.597 μM | SANGER | |||
| SK-N-DZ | Growth Inhibition Assay | IC50=12.0436 μM | SANGER | |||
| NCI-H720 | Growth Inhibition Assay | IC50=12.1705 μM | SANGER | |||
| HCC1187 | Growth Inhibition Assay | IC50=12.2041 μM | SANGER | |||
| IST-SL2 | Growth Inhibition Assay | IC50=12.4872 μM | SANGER | |||
| KE-37 | Growth Inhibition Assay | IC50=12.7966 μM | SANGER | |||
| HCC1599 | Growth Inhibition Assay | IC50=12.9069 μM | SANGER | |||
| A4-Fuk | Growth Inhibition Assay | IC50=12.9586 μM | SANGER | |||
| NKM-1 | Growth Inhibition Assay | IC50=13.2925 μM | SANGER | |||
| BE-13 | Growth Inhibition Assay | IC50=13.7989 μM | SANGER | |||
| MV-4-11 | Growth Inhibition Assay | IC50=14.0324 μM | SANGER | |||
| OPM-2 | Growth Inhibition Assay | IC50=14.4085 μM | SANGER | |||
| KARPAS-422 | Growth Inhibition Assay | IC50=14.5126 μM | SANGER | |||
| RPMI-8226 | Growth Inhibition Assay | IC50=14.8915 μM | SANGER | |||
| KARPAS-45 | Growth Inhibition Assay | IC50=15.7716 μM | SANGER | |||
| SK-PN-DW | Growth Inhibition Assay | IC50=15.8631 μM | SANGER | |||
| LC-2 | Growth Inhibition Assay | IC50=16.1506 μM | SANGER | |||
| NCI-H1648 | Growth Inhibition Assay | IC50=16.254 μM | SANGER | |||
| RL95-2 | Growth Inhibition Assay | IC50=16.3978 μM | SANGER | |||
| KNS-42 | Growth Inhibition Assay | IC50=16.7274 μM | SANGER | |||
| RPMI-6666 | Growth Inhibition Assay | IC50=16.9211 μM | SANGER | |||
| SIG-M5 | Growth Inhibition Assay | IC50=17.1903 μM | SANGER | |||
| VA-ES-BJ | Growth Inhibition Assay | IC50=17.7451 μM | SANGER | |||
| MONO-MAC-6 | Growth Inhibition Assay | IC50=17.9312 μM | SANGER | |||
| LAN-6 | Growth Inhibition Assay | IC50=18.7557 μM | SANGER | |||
| A388 | Growth Inhibition Assay | IC50=19.3059 μM | SANGER | |||
| SK-NEP-1 | Growth Inhibition Assay | IC50=20.2132 μM | SANGER | |||
| TE-10 | Growth Inhibition Assay | IC50=20.5221 μM | SANGER | |||
| HL-60 | Growth Inhibition Assay | IC50=20.9099 μM | SANGER | |||
| MC116 | Growth Inhibition Assay | IC50=21.7221 μM | SANGER | |||
| SW962 | Growth Inhibition Assay | IC50=21.7915 μM | SANGER | |||
| NOMO-1 | Growth Inhibition Assay | IC50=22.6564 μM | SANGER | |||
| CTB-1 | Growth Inhibition Assay | IC50=22.8671 μM | SANGER | |||
| MRK-nu-1 | Growth Inhibition Assay | IC50=22.9074 μM | SANGER | |||
| GR-ST | Growth Inhibition Assay | IC50=23.76 μM | SANGER | |||
| HH | Growth Inhibition Assay | IC50=24.003 μM | SANGER | |||
| NCI-H1963 | Growth Inhibition Assay | IC50=24.0782 μM | SANGER | |||
| QIMR-WIL | Growth Inhibition Assay | IC50=24.8772 μM | SANGER | |||
| CGTH-W-1 | Growth Inhibition Assay | IC50=25.0723 μM | SANGER | |||
| LP-1 | Growth Inhibition Assay | IC50=25.6551 μM | SANGER | |||
| NCI-H748 | Growth Inhibition Assay | IC50=26.5137 μM | SANGER | |||
| PF-382 | Growth Inhibition Assay | IC50=27.2223 μM | SANGER | |||
| ATN-1 | Growth Inhibition Assay | IC50=27.3732 μM | SANGER | |||
| L-540 | Growth Inhibition Assay | IC50=27.6459 μM | SANGER | |||
| LXF-289 | Growth Inhibition Assay | IC50=27.7519 μM | SANGER | |||
| LS-513 | Growth Inhibition Assay | IC50=28.1807 μM | SANGER | |||
| NCI-H1581 | Growth Inhibition Assay | IC50=30.3976 μM | SANGER | |||
| ES6 | Growth Inhibition Assay | IC50=30.6899 μM | SANGER | |||
| SW982 | Growth Inhibition Assay | IC50=30.8566 μM | SANGER | |||
| DOHH-2 | Growth Inhibition Assay | IC50=31.5893 μM | SANGER | |||
| DB | Growth Inhibition Assay | IC50=33.9431 μM | SANGER | |||
| MPP-89 | Growth Inhibition Assay | IC50=34.1756 μM | SANGER | |||
| LB831-BLC | Growth Inhibition Assay | IC50=34.5184 μM | SANGER | |||
| NB5 | Growth Inhibition Assay | IC50=34.8535 μM | SANGER | |||
| GB-1 | Growth Inhibition Assay | IC50=35.0469 μM | SANGER | |||
| TE-15 | Growth Inhibition Assay | IC50=35.2238 μM | SANGER | |||
| LC4-1 | Growth Inhibition Assay | IC50=35.3847 μM | SANGER | |||
| NCI-H747 | Growth Inhibition Assay | IC50=36.1369 μM | SANGER | |||
| NTERA-S-cl-D1 | Growth Inhibition Assay | IC50=38.7347 μM | SANGER | |||
| SK-MM-2 | Growth Inhibition Assay | IC50=40.1146 μM | SANGER | |||
| TGW | Growth Inhibition Assay | IC50=41.0563 μM | SANGER | |||
| ONS-76 | Growth Inhibition Assay | IC50=42.4883 μM | SANGER | |||
| CPC-N | Growth Inhibition Assay | IC50=42.9971 μM | SANGER | |||
| ES4 | Growth Inhibition Assay | IC50=44.4153 μM | SANGER | |||
| Daudi | Growth Inhibition Assay | IC50=45.0827 μM | SANGER | |||
| MOLT-4 | Growth Inhibition Assay | IC50=45.0853 μM | SANGER | |||
| HT-144 | Growth Inhibition Assay | IC50=46.726 μM | SANGER | |||
| SW872 | Growth Inhibition Assay | IC50=48.1933 μM | SANGER | |||
| D-283MED | Growth Inhibition Assay | IC50=48.3542 μM | SANGER | |||
| NCI-H2126 | Growth Inhibition Assay | IC50=48.8476 μM | SANGER | |||
| NCI-SNU-16 | Growth Inhibition Assay | IC50=49.2143 μM | SANGER | |||
| CESS | Growth Inhibition Assay | IC50=49.5088 μM | SANGER | |||
| A101D | Growth Inhibition Assay | IC50=49.9736 μM | SANGER | |||
| Click to View More Cell Line Experimental Data | ||||||
Solubility
|
In vitro |
DMSO
: 9 mg/mL
(19.98 mM)
Water : Insoluble Ethanol : Insoluble |
Molarity Calculator
|
In vivo |
|||||
In vivo Formulation Calculator (Clear solution)
Step 1: Enter information below (Recommended: An additional animal making an allowance for loss during the experiment)
Step 2: Enter the in vivo formulation (This is only the calculator, not formulation. Please contact us first if there is no in vivo formulation at the solubility Section.)
Calculation results:
Working concentration: mg/ml;
Method for preparing DMSO master liquid: mg drug pre-dissolved in μL DMSO ( Master liquid concentration mg/mL, Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug. )
Method for preparing in vivo formulation: Take μL DMSO master liquid, next addμL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O, mix and clarify.
Method for preparing in vivo formulation: Take μL DMSO master liquid, next add μL Corn oil, mix and clarify.
Note: 1. Please make sure the liquid is clear before adding the next solvent.
2. Be sure to add the solvent(s) in order. You must ensure that the solution obtained, in the previous addition, is a clear solution before proceeding to add the next solvent. Physical methods such
as vortex, ultrasound or hot water bath can be used to aid dissolving.
Chemical Information, Storage & Stability
| Molecular Weight | 450.34 | Formula | C21H22Cl2FN5O |
Storage (From the date of receipt) | |
|---|---|---|---|---|---|
| CAS No. | 877399-52-5 | Download SDF | Storage of Stock Solutions |
|
|
| Synonyms | PF-02341066 | Smiles | CC(C1=C(C=CC(=C1Cl)F)Cl)OC2=C(N=CC(=C2)C3=CN(N=C3)C4CCNCC4)N | ||
Mechanism of Action
| Targets/IC50/Ki |
ROS1
(Cell-free assay) <0.025 nM(Ki)
c-Met
(A549, MDA-MB-231, GTL-16, HT29, 786-O, Colo-205, A498 cells) 11 nM
ALK
(Karpas299 cells) 24 nM
|
|---|---|
| In vitro |
PF-2341066 displays similar potency against c-Met phosphorylation in mIMCD3 mouse or MDCK canine epithelial cells with IC50 of 5 nM and 20 nM, respectivly. This compound shows improved or similar activity against NIH3T3 cells engineered to express c-Met ATP-binding site mutants V1092I or H1094R or the P-loop mutant M1250T with IC50 of 19 nM, 2 nM and 15 nM, respectively, compared with NIH3T3 cells expressing wild-type receptor with IC50 of 13 nM. In contrast, a marked shift in potency of this compound is observed against cells engineered to express c-Met activation loop mutants Y1230C and Y1235D with IC50 of 127 nM and 92 nM, respectively, compared with wild-type receptor. It also potently prevents the phosphorylation of c-Met in NCI-H69 and HOP92 cells, with IC50 of 13 nM and 16 nM, respectively, which express the endogenous c-Met variants R988C and T1010I, respectively. This compound is >1,000-fold selective for the VEGFR2 and PDGFRβ RTKs, >250-fold selective for IRK and Lck, and ∼40- to 60-fold selective for Tie2, TrkA, and TrkB, all compared with c-Met. It is 20- to 30-fold selective for RON and Axl RTKs. In contrast, this compound shows a near-equivalent IC50 of 24 nM against the nucleophosmin (NPM)-anaplastic lymphoma kinase (ALK) oncogenic fusion variant of the ALK RTK expressed by the KARPAS299 human anaplastic large cell lymphoma (ALCL) cell line. It inhibits c-Met–dependent neoplastic phenotypes of cancer cells and angiogenic phenotypes of endothelial cells. This chemical suppresses human GTL-16 gastric carcinoma cell growth with IC50 of 9.7 nM. It induces apoptosis in GTL-16 cells with IC50 of 8.4 nM. It inhibits HGF-stimulated human NCI-H441 lung carcinoma cell migration and invasion with IC50 of 11 nM and 6.1 nM, respectively. It inhibits MDCK cell scattering with IC50 of 16 nM. It prevents HGF-stimulated c-Met phosphorylation, cell survival, and Matrigel invasion with IC50 of 11 nM, 14 nM and 35 nM, respectively. In addition, it prevents serum-stimulated HMVEC branching tubulogenesis (formation of vascular tubes) in fibrin gels. It also potently inhibits NPM-ALK phosphorylation in Karpas299 or SU-DHL-1 ALCL cells with an IC50 of 24 nM. This compound potently prevents cell proliferation, which is associated with G(1)-S-phase cell cycle arrest and induction of apoptosis in ALK-positive ALCL cells with IC50 of 30 nM, but not ALK-negative lymphoma cells. Besides, it prevents osteosarcoma behavior associated with primary tumor growth (i.e., proliferation and survival) as well as metastasis (eg, invasion and clonogenicity). |
| Kinase Assay |
Cellular kinase phosphorylation ELISA assays
|
|
Cells are seeded in 96-well plates in media supplemented with 10% fetal bovine serum (FBS) and transferred to serum-free media [with 0.04% bovine serum albumin (BSA)] after 24 h. In experiments investigating ligand-dependent RTK phosphorylation, corresponding growth factors are added for up to 20 min. After incubation of cells with this compound for 1 h and/or appropriate ligands for the designated times, cells are washed once with HBSS supplemented with 1 mM Na3VO4, and protein lysates are generated from cells. Subsequently, phosphorylation of selected protein kinases is assessed by a sandwich ELISA method using specific capture antibodies used to coat 96-well plates and a detection antibody specific for phosphorylated tyrosine residues. Antibody-coated plates are (a) incubated in the presence of protein lysates at 4°C overnight; (b) washed seven times in 1% Tween 20 in PBS; (c) incubated in a horseradish peroxidase–conjugated anti–total-phosphotyrosine (PY-20) antibody (1:500) for 30 min; (d) washed seven times again; (e) incubated in 3,3′,5,5′-tetramethyl benzidine peroxidase substrate to initiate a colorimetric reaction that is stopped by adding 0.09 N H2SO4; and (f) measured for absorbance in 450 nm using a spectrophotometer.
|
|
| In vivo |
In the GTL-16 model, PF-2341066 reveals the ability to cause marked regression of large established tumors (>600 mm3) in both the 50 mg/kg/day and 75 mg/kg/day treatment cohorts, with a 60% decrease in mean tumor volume over the 43-day administration schedule. In an another study, this compound displays the ability to completely inhibits GTL-16 tumor growth for >3 months, with only 1 of 12 mice exhibiting a significant increase in tumor growth over the 3-month treatment schedule at 50 mg/kg/day. In the NCI-H441 NSCLC model, a 43% decrease in mean tumor volume is observed at 50 mg/kg/day during the 38-day PF-2341066 administration cycle. In the Caki-1 RCC model, a 53% decrease in mean tumor volume is observed to be associated with decreased volume of each tumor by at least 30% at 50 mg/kg/day during the 33-day PF-2341066 administration cycle. This compound also reveals near-complete prevention of the growth of established tumors at 50 mg/kg/day in the U87MG glioblastoma or PC-3 prostate carcinoma xenograft models, with 97% or 84% inhibition on the final study day, respectively. In contrast, this chemical p.o. given at 50 mg/kg/day does not significantly inhibit tumor growth in the MDA-MB-231 breast carcinoma model, or the DLD-1 colon carcinoma model. A significant dose-dependent reduction of CD31–positive endothelial cells is observed at 12.5 mg/kg/day, 25 mg/kg/day, and 50 mg/kg/day in GTL-16 tumors, indicating that inhibition of MVD shows a dose-dependent correlation to antitumor efficacy. This compound displays a significant dose-dependent reduction of human VEGFA and IL-8 plasma levels in both the GTL-16 and U87MG models. Marked inhibition of phosphorylated c-Met, Akt, Erk, PLCλ1, and STAT5 levels is observed in GTL-16 tumors following p.o. administration of this compound. P.o. administration of this chemical to severe combined immunodeficient-Beige mice bearing Karpas299 ALCL tumor xenografts leads to dose-dependent antitumor efficacy with complete regression of all tumors at the 100 mg/kg/d dose within 15 days of initial compound administration. In addition, inhibition of key NPM-ALK signaling mediators, including phospholipase C-gamma, signal transducers and activators of transcription 3, extracellular signal-regulated kinases, and Akt by this compound are observed at concentrations or dose levels, which correlated with inhibition of NPM-ALK phosphorylation and function. This compound prevents osteosarcoma behavior associated with primary tumor growth (eg, proliferation and survival) as well as metastasis (eg, invasion and clonogenicity). In nude mice treated with this chemical via oral gavage, the growth and associated osteolysis and extracortical bone matrix formation of osteosarcoma xenografts are prevented by this compound. Treatment of c-MET-amplified GTL-16 xenografts with 50 mg/kg this compound elicits tumor regression that is associated with a slow reduction in 18F-FDG uptake and decreases expression of the glucose transporter 1, GLUT-1. |
References |
|
Applications
| Methods | Biomarkers | Images | PMID |
|---|---|---|---|
| Western blot | pALK / pAKT / pERK / pS6 pROS1 / ROS1 pSTAT3 / STAT3 PARP / cleaved caspase-3 / Bax / Bcl-2 p-c-Met / c-Met p-mTOR |
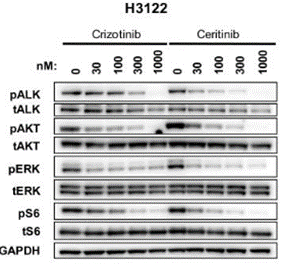
|
24675041 |
| Immunofluorescence | LC3 / lysosome SRC / Met α-tubulin cytochrome c p-ALK |
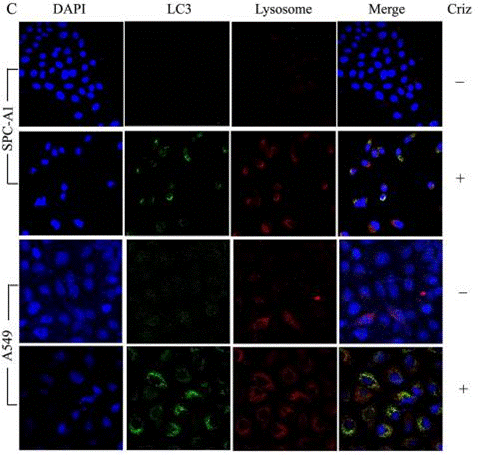
|
26384345 |
Clinical Trial Information
(data from https://clinicaltrials.gov, updated on 2024-05-22)
| NCT Number | Recruitment | Conditions | Sponsor/Collaborators | Start Date | Phases |
|---|---|---|---|---|---|
| NCT06062810 | Not yet recruiting | Non-Small Cell Lung Cancer |
Han Xu M.D. Ph.D. FAPCR Sponsor-Investigator IRB Chair|Medicine Invention Design Inc |
March 28 2024 | Phase 2|Phase 3 |
| NCT04148066 | Completed | Carcinoma Non-Small-Cell Lung |
The Netherlands Cancer Institute|Roche Pharma AG |
July 17 2019 | Not Applicable |
| NCT03947385 | Recruiting | Metastatic Uveal Melanoma|Cutaneous Melanoma|Colorectal Cancer|Other Solid Tumors |
IDEAYA Biosciences |
June 28 2019 | Phase 1|Phase 2 |
| NCT03672643 | Terminated | ALK or ROS1-positive NSCLC |
Pfizer |
January 28 2019 | Phase 4 |
| NCT03439215 | Unknown status | Carcinoma Non-Small-Cell Lung |
Fondazione Ricerca Traslazionale|Clinical research technology Srl |
June 13 2017 | Phase 2 |
Tech Support
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
Frequently Asked Questions
Question 1:
Could you tell me whether this compound represents the pure R-form, or is it the racemic mixture, so a combination of the S- and the R-form?
Answer:
Our S1068 is the R enantiomer (except batches 05 and 06, which are the racemate), and S7505 is the S enantiomer.
Signaling Pathway Map
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.