HDAC inhibitors/activators react with the target HDAC. Histone deacetylases (HDACs) are a family of enzymes that regulate chromatin structure and gene expression by removing acetyl groups from histone proteins. This deacetylation process compacts chromatin, restricting access of transcription factors to DNA and thereby repressing gene transcription. HDAC inhibitors (HDACis), as small-molecule compounds that block HDAC activity, reverse this effect by increasing histone acetylation, leading to chromatin relaxation and the activation of tumor suppressor genes and other functionally important genes. Beyond their role in epigenetics, HDACis also target non-histone proteins, expanding their biological effects across multiple cellular pathways. In recent decades, HDACis have emerged as a pivotal focus in biomedical research, with applications spanning cancer therapy, neurodegenerative disease, and autoimmune disorders.
-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
HDAC Inhibitors/Activators
Other DNA Damage/DNA Repair Related Inhibitors
Isoform-selective Products
| Cat.No. | Product Name | Information | Product Use Citations | Product Validations |
|---|---|---|---|---|
| S2170 | ITF-2357 (Givinostat) Hydrochloride Monohydrate | Givinostat (ITF2357) is a potent HDAC inhibitor for maize HD2, HD1B and HD1A with IC50 of 10 nM, 7.5 nM and 16 nM in cell-free assays. Phase 2. |
|

|
| S2759 | Fimepinostat (CUDC-907) | CUDC-907 is a dual PI3K and HDAC inhibitor for PI3Kα and HDAC1/2/3/10 with IC50 of 19 nM and 1.7 nM/5 nM/1.8 nM/2.8 nM, respectively. CUDC-907 induces cell cycle arrest and apoptosis in breast cancer cells. Phase 1. |
|
|
| S1045 | Trichostatin A (TSA) | TSA (Trichostatin A) is an HDAC inhibitor with IC50 of ~1.8 nM in cell-free assays. |
|

|
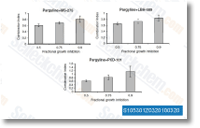
| S1053 | Entinostat (MS-275) | Entinostat (MS-275, SNDX-275) strongly inhibits HDAC1 and HDAC3 with IC50 of 0.51 μM and 1.7 μM in cell-free assays, compared with HDACs 4, 6, 8, and 10. This compound induces autophagy and apoptosis. Phase 3. |
|
|
| S7229 | RGFP966 | RGFP966 is an HDAC3 inhibitor with IC50 of 0.08 μM in cell-free assay, exhibits > 200-fold selectivity over other HDAC. |
|

|
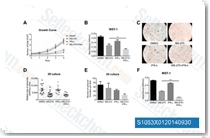
| S1030 | Panobinostat (LBH589) | Panobinostat (LBH589, NVP-LBH589) is a novel broad-spectrum HDAC inhibitor with IC50 of 5 nM in a cell-free assay. It induces autophagy and apoptosis, and effectively disrupts HIV latency in vivo. Phase 3. |
|
|
| S1085 | Belinostat (PXD101) | Belinostat is a novel HDAC inhibitor with IC50 of 27 nM in a cell-free assay, with activity demonstrated in cisplatin-resistant tumors. Belinostat (PXD101) induces autophagy. |
|

|
| S1047 | Vorinostat (SAHA) | Vorinostat (SAHA) is an HDAC inhibitor with IC50 of ~10 nM in a cell-free assay and abrogates productive HPV-18 DNA amplification. |
|

|
| S3020 | Romidepsin (FK228) | Romidepsin (FK228, Depsipeptide, FR 901228, NSC 630176) is a potent HDAC1 and HDAC2 inhibitor with IC50 of 36 nM and 47 nM in cell-free assays, respectively. This compound controls growth and induces apoptosis in neuroblastoma tumor cells. |
|

|
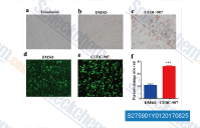
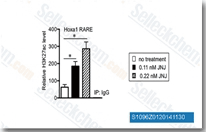
| S1096 | Quisinostat (JNJ-26481585) Dihydrochloride | Quisinostat (JNJ-26481585) 2HCl is a novel second-generation HDAC inhibitor with highest potency for HDAC1 with IC50 of 0.11 nM in a cell-free assay, modest potent to HDACs 2, 4, 10, and 11; greater than 30-fold selectivity against HDACs 3, 5, 8, and 9 and lowest potency to HDACs 6 and 7. Phase 2. |
|
|
Signaling Pathway Map
Classification of HDACs and HDAC Inhibitors
To understand HDACis, it is first critical to categorize their targets: HDACs are divided into four classes based on sequence homology and cofactor requirements. Class I HDACs (HDAC1, 2, 3, 8) are primarily localized in the nucleus and are ubiquitously expressed, playing essential roles in cell cycle regulation and proliferation. Class II HDACs (subdivided into IIa: HDAC4, 5, 7, 9; IIb: HDAC6, 10) shuttle between the nucleus and cytoplasm, with Class IIa involved in developmental signaling and Class IIb regulating cytoskeletal dynamics and protein degradation. Class III HDACs, also called sirtuins (SIRT1–7), depend on NAD+ as a cofactor and are implicated in metabolism, aging, and stress responses. Class IV HDAC (HDAC11) is the smallest family member, with functions in immune cell activation and lipid metabolism. HDACis are correspondingly classified based on their chemical structure and HDAC class selectivity:
1.1 Pan-HDACis:
Inhibit multiple HDAC classes, particularly Class I and II. Examples include vorinostat (SAHA) and trichostatin A (TSA). Vorinostat, the first FDA-approved HDACi (2006), targets Class I (HDAC1, 2, 3) and Class IIb (HDAC6) and is used to treat cutaneous T-cell lymphoma (CTCL).
1.2 Class I-Selective HDACis:
Preferentially inhibit Class I HDACs, reducing off-target effects on Class II enzymes. Romidepsin, a cyclic peptide HDACi, selectively targets HDAC1 and 2 and is approved for relapsed CTCL and peripheral T-cell lymphoma (PTCL).
1.3 Class II-Selective HDACis:
Focus on Class II HDACs, with potential applications in neurodegeneration and cardiovascular disease. For example, MC1568 inhibits Class IIa HDACs (HDAC4, 5, 7) and has shown promise in preclinical models of Huntington’s disease.
1.4 Class III HDAC (Sirtuin) Modulators:
Unlike other HDACis, sirtuin-targeting compounds often act as activators (e.g., resveratrol, which activates SIRT1) due to the unique NAD+-dependent mechanism of sirtuins. These modulators are studied for their roles in aging and metabolic disorders.
2. Mechanisms of Action: Beyond Histone Acetylation
The canonical mechanism of HDACis involves histone acetylation, but their biological effects extend far beyond this epigenetic regulation, driven by the modulation of non-histone proteins.
2.1 Histone-Mediated Epigenetic Regulation
HDACs remove acetyl groups from the ε-amino groups of lysine residues on histone tails (e.g., H3K9, H3K14, H4K8). This deacetylation increases the positive charge of histones, strengthening their electrostatic interaction with negatively charged DNA and forming a condensed chromatin structure (heterochromatin) that blocks transcription. HDACis bind to the catalytic site of HDACs, preventing deacetylation and accumulating acetylated histones. This relaxed chromatin (euchromatin) allows transcription factors, such as p53 and E2F, to bind to promoter regions, activating the expression of tumor suppressor genes (e.g., p21 Bax) and genes involved in cell cycle arrest and apoptosis. In cancer cells, this reactivation reverses the oncogenic silencing of critical pathways, suppressing proliferation and inducing cell death.
2.2 Regulation of Non-Histone Substrates
Non-histone proteins constitute a large fraction of HDAC substrates, and their acetylation status controls protein function, localization, and stability. HDACis modulate key non-histone targets, including:
p53: A master tumor suppressor. Acetylation of p53 (at lysine residues 373 and 382) by HDAC inhibition enhances its stability and DNA-binding activity, promoting cell cycle arrest and apoptosis in cancer cells.
NF-κB: A transcription factor central to inflammation and immune responses. HDACs deacetylate the p65 subunit of NF-κB, enhancing its transcriptional activity. HDACis block this deacetylation, inhibiting NF-κB-mediated expression of pro-inflammatory cytokines (e.g., TNF-α, IL-6) and reducing tumor cell survival.
α-Tubulin: A component of the cytoskeleton. HDAC6 (a Class IIb HDAC) deacetylates α-tubulin, regulating microtubule dynamics. HDACis targeting HDAC6 increase α-tubulin acetylation, disrupting mitosis and inhibiting cancer cell migration and invasion.
These non-histone effects expand the therapeutic potential of HDACis beyond cancer, as seen in their ability to reduce inflammation in autoimmune diseases and protect neurons in neurodegenerative disorders.
3. Key Research Advances in HDAC Inhibitor Development
Recent research has advanced HDACi development in three major directions: optimizing efficacy in cancer therapy, exploring applications in non-oncological diseases, and improving selectivity to reduce side effects.
3.1 Cancer Therapy: From Monotherapy to Combination Strategies
While single-agent HDACis have shown efficacy in hematological malignancies (e.g., CTCL, PTCL), their performance in solid tumors has been limited due to tumor heterogeneity and drug resistance. To address this, researchers have focused on combination therapies that synergize with HDACis:
Immune Checkpoint Inhibitors: HDAC is modulate the tumor microenvironment by increasing the expression of tumor-associated antigens (TAAs) and MHC class I molecules, enhancing T-cell recognition of cancer cells. Preclinical studies have shown that combining vorinostat with anti-PD-1 antibodies (e.g., pembrolizumab) improves T-cell infiltration and tumor regression in melanoma and non-small cell lung cancer (NSCLC). A 2023 clinical trial (NCT03123096) reported that the combination of romidepsin and (e.g., nivolumab) achieved a 35% objective response rate in relapsed PTCL, significantly higher than romidepsin monotherapy (15%).
Targeted Therapies: HDACis synergize with inhibitors of oncogenic pathways, such as the PI3K/Akt/mTOR pathway. In breast cancer models, combining the Class I HDACi entinostat with the PI3K inhibitor (e.g., alpelisib) enhances apoptosis by blocking mTOR-mediated survival signals. Similarly, in glioblastoma, HDACis reverse the resistance to EGFR inhibitors by reactivating silenced tumor suppressor genes (e.g., PTEN).
3.2 Non-Oncological Applications: Neurodegeneration and Autoimmunity
HDACis have gained traction in non-cancer research, particularly in neurodegenerative diseases where epigenetic dysregulation contributes to pathogenesis:
3.2.1 Alzheimer’s Disease (AD): HDACis increase the acetylation of histones and tau protein (a key component of neurofibrillary tangles in AD). Preclinical studies with vorinostat and panobinostat have shown reduced tau hyperphosphorylation and improved cognitive function in AD mouse models. A 2022 study in Nature Communications reported that a Class IIa HDACi (HDAC4/5 inhibitor) enhances the clearance of amyloid-beta (Aβ) plaques by activating microglia, a critical step in reducing neuroinflammation in AD.
3.2.2 Autoimmune Diseases: HDACis suppress immune cell activation by inhibiting NF-κB and reducing cytokine production. In rheumatoid arthritis (RA), HDACis (e.g., givinostat) have been tested in phase II trials, showing reduced joint inflammation and improved disease activity scores by inhibiting the proliferation of synovial fibroblasts and the production of IL-6 and TNF-α.
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.