STAT inhibitors are a class of targeted biological agents designed to modulate the signal transducer and activator of transcription (STAT) signaling pathway, a central cascade in regulating cellular processes such as proliferation, differentiation, apoptosis, and immune response. Dysregulation of STAT signaling is closely associated with the pathogenesis of numerous diseases, including various cancers, autoimmune disorders, and inflammatory conditions, making STAT inhibitors a focal point of contemporary biomedical research. This article focuses on the scientific research advances of STAT inhibitors, exploring their mechanism of action, research progress in specific disease fields, and current challenges and prospects.
-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
STAT Inhibitors/Modulators
| Cat.No. | Product Name | Information | Product Use Citations | Product Validations |
|---|---|---|---|---|
| S7977 | Napabucasin (BBI608) | Napabucasin (BBI608) is an orally available Stat3 and cancer cell stemness inhibitor. |
|
|
| S1491 | Fludarabine | Fludarabine is a STAT1 activation inhibitor which causes a specific depletion of STAT1 protein (and mRNA) but not of other STATs. Also a DNA synthesis inhibitor in vascular smooth muscle cells. Fludarabine induces apoptosis. |
|
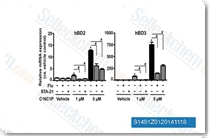
|
| S1229 | Fludarabine (NSC 118218) Phosphate | Fludarabine Phosphate is an analogue of adenosine and deoxyadenosine, which is able to compete with dATP for incorporation into DNA and inhibit DNA synthesis. |
|
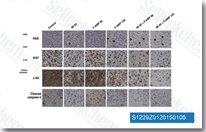
|
| S1396 | Resveratrol (trans-Resveratrol) | Resveratrol has a wide spectrum of targets including cyclooxygenases(i.e. COX, IC50=1.1 μM), lipooxygenases(LOX, IC50=2.7 μM), kinases, sirtuins and other proteins. It has anti-cancer, anti-inflammatory, blood-sugar-lowering and other beneficial cardiovascular effects. Resveratrol induces mitophagy/autophagy and autophagy-dependent apoptosis. |
|
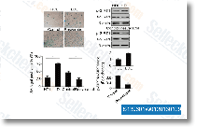
|
| S7024 | Stattic | Stattic, the first nonpeptidic small molecule, potently inhibits STAT3 activation and nuclear translocation with IC50 of 5.1 μM in cell-free assays, highly selectivity over STAT1. Stattic induces apoptosis. |
|

|
| S1155 | NSC 74859 (S3I-201) | NSC 74859 (S3I-201) shows potent inhibition of STAT3 DNA-binding activity with IC50 of 86 μM in cell-free assays, and low activity towards STAT1 and STAT5. |
|

|
| S2285 | Cryptotanshinone (Tanshinone C) | Cryptotanshinone (Tanshinone C) is a STAT3 inhibitor with IC50 of 4.6 μM in a cell-free assay, strongly inhibiting phosphorylation of STAT3 Tyr705 with a small effect on STAT3 Ser727, but none against STAT1 nor STAT5. It also induces ROS-dependent autophagy and mitochondria-mediated apoptosis. |
|
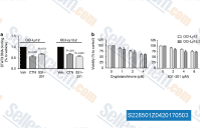
|
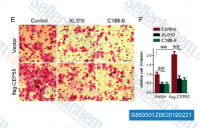
| S8605 | C188-9 (TTI-101) | C188-9 (TTI 101) is a potent inhibitor of STAT3 that binds to STAT3 with high affinity (KD=4.7±0.4 nM). C188-9 is well tolerated in mice, shows good oral bioavailability, and is concentrated in tumors. |
|
|
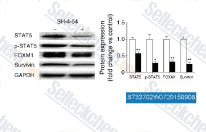
| S7337 | SH-4-54 | SH-4-54 is a potent STAT inhibitor with KD of 300 nM and 464 nM for STAT3 and STAT5, respectively. |
|
|
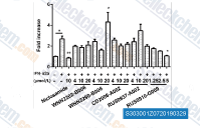
| S3030 | Niclosamide | Niclosamide can inhibit DNA replication and inhibit STAT3 with IC50 of 0.7 μM in a cell-free assay. This compound selectively inhibited the phosphorylation of STAT3 and had no obvious inhibition against the activation of other homologues (e.g., STAT1 and STAT5). |
|
|
Signaling Pathway Map
Mechanistic Basis: Targeting the JAK-STAT Pathway and Key Kinases
The core mechanism of STAT inhibitors lies in their ability to interfere with the aberrant activation of the JAK-STAT pathway, which is initiated by cytokine or growth factor binding to cell surface receptors. This binding induces the activation of Janus kinases (JAKs), a family of non-receptor tyrosine kinases that phosphorylate STAT proteins. Phosphorylated STATs dimerize, translocate to the nucleus, and bind to specific DNA response elements to regulate gene expression. Dysregulation of this pathway, often driven by mutations or overexpression of JAKs, STATs, or upstream receptors, leads to uncontrolled cellular proliferation and immune dysfunction—hallmarks of many diseases.
Kinase-Targeted STAT Inhibitors: Focus on JAK Inhibition
Kinases, particularly JAKs, are critical upstream regulators of STAT activation, making them primary targets for STAT pathway modulation. JAK inhibitors (JAKi), a subset of STAT inhibitors, act by competing with ATP for the kinase domain of JAKs, thereby inhibiting JAK-mediated STAT phosphorylation. Early research identified four JAK isoforms (JAK1, JAK2, JAK3, Tyk2), each with distinct tissue expression and cytokine specificity. For example, JAK2 mutations are prevalent in myeloproliferative neoplasms (MPNs), and JAK2-specific inhibitors have shown efficacy in reducing splenomegaly and symptom burden in MPN patients. Pan-JAK inhibitors, such as tofacitinib, which targets JAK1/3, have been approved for autoimmune diseases, highlighting the clinical potential of kinase-targeted STAT inhibition. However, off-target kinase inhibition remains a challenge, often leading to adverse effects such as myelosuppression and increased infection risk.
Direct STAT-Targeted Inhibitors: Overcoming Kinase Redundancy
To address the limitations of kinase-targeted inhibitors, research has shifted toward developing direct STAT inhibitors that target STAT proteins themselves, bypassing upstream kinase redundancy. These inhibitors act through multiple mechanisms: blocking STAT phosphorylation, interfering with dimerization, or inhibiting nuclear translocation and DNA binding. For instance, small-molecule inhibitors targeting the SH2 domain of STATs—critical for both phosphorylation and dimerization—have been identified. Preclinical studies show that these inhibitors can selectively suppress STAT activation without affecting other kinase pathways, reducing off-target effects. Direct STAT inhibitors also offer the advantage of targeting specific STAT isoforms, allowing for tailored therapy based on disease-specific STAT dysregulation.
STAT Inhibitors in Cancer Research: Targeting STAT1 and Beyond
Cancer is one of the most extensively studied fields for STAT inhibitors, given the frequent dysregulation of the JAK-STAT pathway in malignant tumors. Different STAT isoforms play distinct roles in cancer: while STAT3 and STAT5 are often oncogenic, promoting tumor cell proliferation and angiogenesis, STAT1 exhibits context-dependent functions—acting as a tumor suppressor in some cancers (e.g., colorectal cancer) by inducing apoptosis and immune surveillance, but as an oncogene in others (e.g., melanoma) by mediating treatment resistance.
STAT1-Targeted Strategies in Cancer Therapy
The dual role of STAT1 in cancer presents unique challenges and opportunities for inhibitor development. In cancers where STAT1 is oncogenic, such as advanced melanoma, STAT1 inhibitors have shown promise in reversing resistance to immune checkpoint inhibitors. Preclinical models demonstrate that inhibiting STAT1 phosphorylation reduces the expression of immune-suppressive molecules (e.g., PD-L1) on tumor cells, enhancing T-cell infiltration and anti-tumor immunity. Conversely, in cancers where STAT1 is a tumor suppressor, indirect STAT modulation—such as activating STAT1 through JAK inhibition in specific contexts—has been explored. For example, in some leukemia models, low-dose JAK inhibitors can upregulate STAT1 expression, inducing tumor cell apoptosis. These findings emphasize the need for personalized approaches based on STAT1 status in individual tumors.
Broad-Spectrum STAT Inhibitors in Solid and Hematological Cancers
Beyond STAT1, STAT inhibitors targeting other isoforms have advanced to clinical trials. For solid cancers, STAT3 inhibitors have shown efficacy in preclinical models of breast, lung, and pancreatic cancer, where STAT3 is frequently overactivated. One such inhibitor, napabucasin, targets STAT3 nuclear translocation and has entered phase III trials for colorectal cancer. In hematological cancers, JAK2 inhibitors (e.g., ruxolitinib) are already approved for MPNs, and STAT5 inhibitors are being developed for chronic myeloid leukemia (CML) and T-cell acute lymphoblastic leukemia (T-ALL), where STAT5 drives leukemic cell survival. Combination therapy—pairing STAT inhibitors with chemotherapy, targeted agents, or immunotherapies—is a growing area of research, aiming to overcome resistance and enhance efficacy.
STAT Inhibitors in Dermatology: Addressing Inflammatory and Neoplastic Conditions
The JAK-STAT pathway plays a pivotal role in the pathogenesis of various dermatological diseases, including inflammatory disorders (e.g., psoriasis, atopic dermatitis) and cutaneous malignancies (e.g., cutaneous T-cell lymphoma, CTCL). This has positioned STAT inhibitors as promising therapeutic agents in dermatology research.
Inflammatory Skin Diseases: Targeting JAK-STAT-Mediated Inflammation
Psoriasis and atopic dermatitis are characterized by excessive inflammation driven by cytokines (e.g., IL-23, IL-17, IL-4) that signal through the JAK-STAT pathway. Topical and systemic JAK inhibitors have emerged as effective treatments. For example, topical tofacitinib and ruxolitinib have been approved for mild-to-moderate atopic dermatitis and psoriasis, respectively, by suppressing STAT3 and STAT6 activation, which reduce the production of inflammatory cytokines. Clinical trials show that these inhibitors achieve rapid symptom relief (e.g., itching, erythema) with manageable side effects compared to traditional therapies like corticosteroids. Research is now focused on developing isoform-specific JAK inhibitors (e.g., TYK2 inhibitors) to further improve safety and efficacy.
Cutaneous Malignancies: STAT Inhibitors as Therapeutic Agents
Cutaneous malignancies, such as CTCL and melanoma, often exhibit dysregulated JAK-STAT signaling. In CTCL, a non-Hodgkin lymphoma of skin-homing T cells, STAT3 and STAT5 are overactivated, promoting T-cell proliferation and immune evasion. Systemic JAK inhibitors (e.g., ruxolitinib) have shown clinical benefit in refractory CTCL, reducing skin lesions and improving quality of life. In melanoma, as mentioned earlier, STAT1 inhibitors can enhance the efficacy of immune checkpoint inhibitors by reversing immune suppression. Additionally, topical STAT inhibitors are being explored for pre-malignant conditions like actinic keratosis, aiming to prevent progression to squamous cell carcinoma. These applications highlight the versatility of STAT inhibitors in dermatological research. In conclusion, STAT inhibitors represent a diverse and promising class of therapeutic agents with extensive research applications across oncology, dermatology, and beyond. While kinase-targeted inhibitors (e.g., JAK inhibitors) have made significant clinical strides, direct STAT inhibitors and personalized combination strategies hold the key to overcoming current limitations. Future research will focus on improving specificity, reducing toxicity, and identifying predictive biomarkers to maximize the clinical potential of STAT inhibitors in various disease contexts.
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.