-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
research use only
Fenebrutinib (GDC-0853) BTK inhibitor
Cat.No.S8421
Chemical Structure
Molecular Weight: 664.80
Quality Control
| Related Targets | EGFR VEGFR FGFR PDGFR c-Met Src MEK CSF-1R FLT3 HER2 |
|---|---|
| Other BTK Inhibitors | Catadegbrutinib (BGB-16673) Spebrutinib (AVL-292) tirabrutinib(ONO-4059) hydrochloride LFM-A13 CGI1746 Evobrutinib BMS-935177 CNX-774 Branebrutinib (BMS-986195) Pirtobrutinib (LOXO-305) |
Solubility
|
In vitro |
DMSO
: 8 mg/mL
(12.03 mM)
Water : Insoluble Ethanol : Insoluble |
Molarity Calculator
|
In vivo |
|||||
In vivo Formulation Calculator (Clear solution)
Step 1: Enter information below (Recommended: An additional animal making an allowance for loss during the experiment)
Step 2: Enter the in vivo formulation (This is only the calculator, not formulation. Please contact us first if there is no in vivo formulation at the solubility Section.)
Calculation results:
Working concentration: mg/ml;
Method for preparing DMSO master liquid: mg drug pre-dissolved in μL DMSO ( Master liquid concentration mg/mL, Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug. )
Method for preparing in vivo formulation: Take μL DMSO master liquid, next addμL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O, mix and clarify.
Method for preparing in vivo formulation: Take μL DMSO master liquid, next add μL Corn oil, mix and clarify.
Note: 1. Please make sure the liquid is clear before adding the next solvent.
2. Be sure to add the solvent(s) in order. You must ensure that the solution obtained, in the previous addition, is a clear solution before proceeding to add the next solvent. Physical methods such
as vortex, ultrasound or hot water bath can be used to aid dissolving.
Chemical Information, Storage & Stability
| Molecular Weight | 664.80 | Formula | C37H44N8O4 |
Storage (From the date of receipt) | 3 years -20°C powder |
|---|---|---|---|---|---|
| CAS No. | 1434048-34-6 | -- | Storage of Stock Solutions |
|
|
| Synonyms | N/A | Smiles | CC1CN(CCN1C2=CN=C(C=C2)NC3=CC(=CN(C3=O)C)C4=C(C(=NC=C4)N5CCN6C7=C(CC(C7)(C)C)C=C6C5=O)CO)C8COC8 | ||
Mechanism of Action
| Targets/IC50/Ki |
BTK
(Cell-free assay) 0.91 nM(Ki)
BTK C481R
(Cell-free assay) 1.3 nM(Ki)
BTK C481S
(Cell-free assay) 1.6 nM(Ki)
BTK T474M
(Cell-free assay) 3.4 nM(Ki)
BTK T474I
(Cell-free assay) 12.6 nM(Ki)
|
|---|---|
| In vitro |
When tested at 1 μM against a broad panel of human kinase biochemical assays, Fenebrutinib (GDC-0853) inhibits only 3 of 286 off-target kinases. Based on the determined IC50 values, the selectivity for Btk is >100-fold against each of these 3 off-targets: Bmx (153-fold), Fgr (168-fold), and Src (131-fold). This compound blocks both B-cell BCR and monocyte FcγR signaling. In in vitro biochemical Btk enzyme assay, it displays an average residence time with Btk of 18.3 ± 2.8 hours. It blocks cellular autophosphorylation of WT Btk and the C481S mutant. CLL (chronic lymphocytic leukemia) cells treated with GDC-0853 in vitro before BCR stimulation demonstrate reduced levels of BTK phosphorylation and diminished activation of downstream targets including PLCγ2, AKT, and ERK. It inhibits NF-κB–dependent transcription, reduces activation, and impairs migration. This compound lacks inhibition of EGFR and ITK in cellular system and does not affect T-cell receptor activation. |
| Kinase Assay |
Kinase selectivity
|
|
Fenebrutinib (GDC-0853) kinase selectivity is assessed at a concentration of 1 µM in a panel of up to 287 recombinant human kinase activity and binding assays, including cytoplasmic and receptor tyrosine kinases, serine/threonine kinases, and lipid kinases. The kinase activity assays measure peptide phosphorylation or ADP production while the binding assays monitored displacement of ATP sitebinding probes. The ATP concentrations used in the activity assays are typically within 2-fold of the experimentally determined apparent Michaelis constant (Kmapp) value for each kinase while the competitive binding tracer concentrations used in the binding assays are generally within 3-fold of the experimentally determined dissociation constant (Kd) values. This compound is tested in duplicate against each kinase and the mean % Inhibition values are reported. For kinases that are inhibited by close to or greater than 80% at the test concentration, 10-point inhibitor titrations using the same assays are carried out in order to determine the inhibitor concentrations that caused 50% inhibition (IC50).
|
|
| In vivo |
Fenebrutinib (GDC-0853) has moderate clearance of 27.4 mL/min/kg and excellent bioavailability (F=65%) in rats administered 0.2 mg/kg via intraperitoneal injection or 1 mg/kg PO. Its plasma clearance is 27.4 mL/min/kg, the volume of distribution (Vd) is 5.42 L/kg and the plasma half-life (t1/2) is 2.2 h. This compound also demonstrates favorable PK properties in dogs. The 3.8-hour half-life (Clp 10.9 mL/min/kg, Vd 2.96 L/kg) and high oral bioavailability (85%) also enable attainment of sufficient exposures in dog toxicology studies. It is well tolerated in both rats and dogs and displays an overall favorable safety profile. GDC-0853 is useful in treating rheumatoid arthritis and other B-cell or myeloid cell mediated autoimmune diseases. In a single ascending dose (SAD) study (0.5 mg to 600 mg) and multiple ascending dose (MAD) study for 14 days (250 mg BID to 500 mg QD), it is very well tolerated with no severe adverse events, no safety signals, and no dose limiting toxicities. It is well absorbed and had linear, doseproportional pharmacokinetics. In Sprague-Dawley (SD) rats, administration of this compound and other structurally diverse BTK inhibitors for 7 days or longer cause pancreatic lesions consisting of multifocal islet-centered hemorrhage, inflammation, fibrosis, and pigment-laden macrophages with adjacent lobular exocrine acinar cell atrophy, degeneration, and inflammation. Similar findings are not observed in mice or dogs at much higher exposures. |
References |
|
Applications
| Methods | Biomarkers | Images | PMID |
|---|---|---|---|
| Western blot | p-BTK / BTK / p-PLCγ2 / PLCγ2 / p-AKT / AKT / p-ERK / ERK |
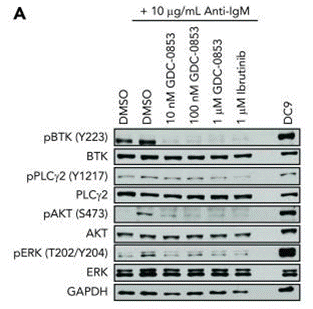
|
30018078 |
Clinical Trial Information
(data from https://clinicaltrials.gov, updated on 2024-05-22)
| NCT Number | Recruitment | Conditions | Sponsor/Collaborators | Start Date | Phases |
|---|---|---|---|---|---|
| NCT05119569 | Active not recruiting | Relapsing Multiple Sclerosis |
Hoffmann-La Roche |
March 1 2022 | Phase 2 |
| NCT04586023 | Active not recruiting | Relapsing Multiple Sclerosis |
Hoffmann-La Roche |
March 24 2021 | Phase 3 |
| NCT04586010 | Active not recruiting | Relapsing Multiple Sclerosis |
Hoffmann-La Roche |
March 17 2021 | Phase 3 |
| NCT03693625 | Terminated | Urticaria |
Genentech Inc. |
September 27 2018 | Phase 2 |
| NCT03596632 | Completed | Healthy Participants |
Hoffmann-La Roche |
July 27 2018 | Phase 1 |
Tech Support
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
Signaling Pathway Map
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.