-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
research use only
Tariquidar (XR9576) P-gp inhibitor
Cat.No.S8028
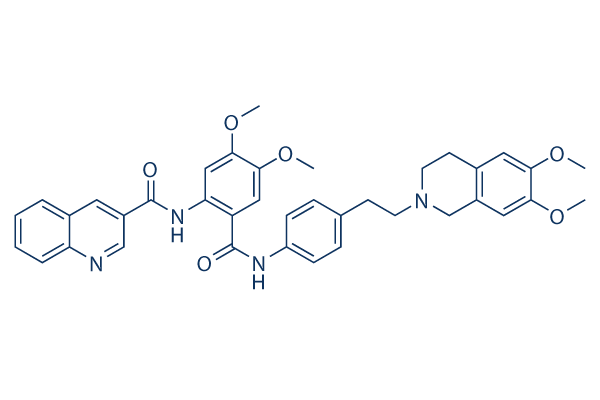
Chemical Structure
Molecular Weight: 646.73
Quality Control
Cell Culture, Treatment & Working Concentration
| Cell Lines | Assay Type | Concentration | Incubation Time | Formulation | Activity Description | PMID |
|---|---|---|---|---|---|---|
| K562/DOX | Function assay | 1 uM | 10 mins | Inhibition of P-gp in human K562/DOX cells assessed as increase in rhodamine-123 efflux in human K562 cells at 1 uM incubated for 10 mins hrs by flow cytometry relative to untreated control | 28113128 | |
| NB1643 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for NB1643 cells | 29435139 | |||
| SK-N-SH | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SK-N-SH cells | 29435139 | |||
| KB-V1 | Function assay | 200 nM | Inhibition of P-gp in human KB-V1 cells assessed as increase in rhodamine 123 accumulation at 200 nM | 21657271 | ||
| SK-N-MC | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SK-N-MC cells | 29435139 | |||
| HepG2 | Cytotoxicity assay | 48 hrs | Cytotoxicity against adriamycin-resistant human HepG2 cells assessed as inhibition of cell proliferation after 48 hrs by MTT assay, IC50=37.2μM | 27328029 | ||
| K562 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human K562 cells after 48 hrs by MTT assay, IC50=31.56μM | 28645831 | ||
| K562 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human K562 cells assessed as reduction in cell viability after 48 hrs by MTT assay, IC50=31.56μM | 29631786 | ||
| A2780adr | Function assay | 10 uM | 30 mins | Inhibition of ABCB1 in human A2780adr cells assessed as increase in accumulation of calcein AM at 10 uM preincubated for 30 mins followed by calcein AM addition measured every 60 secs for 60 mins by fluorescence assay relative to control | 29547272 | |
| K562/A02 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human K562/A02 cells overexpressing P-gp assessed as reduction in cell viability after 48 hrs by MTT assay, IC50=27.19μM | 29631786 | ||
| CCD-18Co | Cytotoxicity assay | 48 hrs | Cytotoxicity against human CCD-18Co cells assessed as cell viability after 48 hrs by MTT assay, IC50=25μM | 26197160 | ||
| SW620/AD300 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human SW620/AD300 cells assessed as cell viability after 48 hrs by MTT assay, IC50=25μM | 26197160 | ||
| HLF | Cytotoxicity assay | 48 hrs | Cytotoxicity against HLF cells assessed as inhibition of cell proliferation after 48 hrs by MTT assay, IC50=16.69μM | 27328029 | ||
| CEM/VLB500 | Growth inhibition assay | 3 days | Growth inhibition of human CEM/VLB500 cells after 3 days by resazurin assay, GI50=13.5μM | 17399990 | ||
| MCF7/ADR | Cytotoxicity assay | 48 hrs | Intrinsic cytotoxicity against human MCF7/ADR cells assessed as inhibition of cell proliferation after 48 hrs by MTT assay, IC50=13.1μM | 27328029 | ||
| HCT116 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human HCT116 cells assessed as cell viability after 48 hrs by MTT assay, IC50=12.5μM | 26197160 | ||
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 treated for 48 hrs followed by compound washout measured after 12 hrs by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=8.28μM | 28645831 | ||
| KBV | Function assay | 5 uM | 72 hrs | Reversal of P-gp-mediated drug resistance in human KBV cells assessed as potentiation of cytotoxicity by measuring IC50 at 5 uM after 72 hrs by MTT assay (Rvb = 398.34 +/- 0.58 uM), IC50=5.24μM | 30384042 | |
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 treated for 48 hrs followed by compound washout measured after 6 hrs by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=4.97μM | 28645831 | ||
| KBV | Function assay | 10 uM | 72 hrs | Reversal of P-gp-mediated drug resistance in human KBV cells assessed as potentiation of cytotoxicity by measuring IC50 at 10 uM after 72 hrs by MTT assay (Rvb = 398.34 +/- 0.58 uM), IC50=4.46μM | 30384042 | |
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 treated for 48 hrs followed by compound washout measured immediately by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=3.02μM | 28645831 | ||
| K562/A02 | Function assay | 5 uM | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 at 5 uM measured after 48 hrs by MTT assay (Rvb = 43.75 to 96.91 uM), IC50=1.97μM | 28645831 | |
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 measured after 48 hrs by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=1.6μM | 28645831 | ||
| MCF-7 MX | Function assay | Inhibition of BCRP expressed in MCF-7 MX cells using Hoechst 33342 staining, IC50=1.5μM | 21354800 | |||
| MCF7 | Function assay | Inhibition of ABCG2 in human mitoxantrone-resistant MCF7 cells by Hoechst 33342 assay, IC50=1.44544μM | 18678495 | |||
| HFE | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HFE cells assessed as cell viability after 72 hrs by MTT assay, IC50=1.28μM | 26197160 | ||
| MDCK | Function assay | Inhibition of BCRP expressed in MDCK cells using Hoechst 33342 staining, IC50=0.94μM | 21354800 | |||
| MCF7/Topo | Function assay | Inhibition of ABCG2 overexpressed in human MCF7/Topo cells by flow cytometric-based mitoxantrone efflux assay, IC50=0.916μM | 19170519 | |||
| MCF7/Topo | Function assay | 2 hrs | Inhibition of ABCG2 in human MCF7/Topo cells after 2 hrs by Hoechst 33342 staining based fluorescence assay, IC50=0.526μM | 30128080 | ||
| MCF7/Topo | Function assay | 2 hrs | Inhibition of ABCG2 in human MCF7/Topo cells after 2 hrs by Hoechst 33342 microplate assay, IC50=0.526μM | 24900683 | ||
| MCF7/Topo | Function assay | Inhibition of ABCG2 expressed in human MCF7/Topo cells by Hoechst microplate assay, IC50=0.526μM | 21570282 | |||
| MCF7/Topo | Function assay | Inhibition of ABCG2 in human MCF7/Topo cells by Hoechst 33342 assay, IC50=0.52μM | 26774038 | |||
| KBV1 | Function assay | 10 mins | Inhibition of ABCB1 in human KBV1 cells after 10 mins by Calcein-AM microplate assay, IC50=0.223μM | 24900683 | ||
| Kb-V1 | Function assay | 10 mins | Inhibition of ABCB1 expressed in Kb-V1 cells after 10 mins by calcein-AM assay, IC50=0.223μM | 21570282 | ||
| KBv1 | Function assay | Inhibition of ABCB1 overexpressed in human KBv1 cells by flow cytometric-based calcein-AM efflux assay, IC50=0.223μM | 19170519 | |||
| KBV1 | Function assay | Inhibition of ABCB1 in human KBV1 cells assessed as inhibition of calcein-AM efflux, IC50=0.22μM | 26774038 | |||
| A2780 | Function assay | 30 mins | Inhibition of human Pgp in A2780 cells after 30 mins by Hoechst 33342 assay, IC50=0.12589μM | 18083034 | ||
| KB-3-1 | Function assay | 1000 nM | 72 hrs | Potentiation of doxorubicin-induced cytotoxicity against human KB-3-1 cells assessed as doxorubicin IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 0.15 +/- 0.04 uM), IC50=0.11μM | 27504669 | |
| OVCAR8 | Function assay | 1000 nM | 72 hrs | Potentiation of doxorubicin-induced cytotoxicity against human OVCAR8 cells assessed as doxorubicin IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 0.12 +/- 0.03 uM), IC50=0.08μM | 27504669 | |
| A2780/ADR | Function assay | Inhibition of P-glycoprotein-mediated multidrug resistance in adriamycin-resistant human A2780/ADR cells by calcein AM assay, IC50=0.078μM | 19250834 | |||
| A2780adr | Function assay | Inhibition of P-gp expressed in A2780adr cells by calcein AM accumulation assay, IC50=0.08μM | 21354800 | |||
| A2780 | Function assay | Inhibition of P-gp in human adriamycin-resistant A2780 cells by Hoechst 33342 assay, IC50=0.07244μM | 18678495 | |||
| KBV1 | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 expressed in KBV1 cells assessed as potentiation of doxorubicin-induced cytotoxicity by measuring doxorubicin IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 5.07 +/- 0.19 uM), IC50=0.07μM | 27504669 | |
| CEM/VLB500 | Function assay | 3 days | Reversal of P-gp-mediated multidrug resistance to in human CEM/VLB500 cells after 3 days by resazurin assay, EC50=0.068μM | 17399990 | ||
| EMT6/AR1.0 | Function assay | 1 hr | Inhibition of mouse Pgp in EMT6/AR1.0 cells after 1 hr by daunorubicin accumulation assay, IC50=0.06457μM | 18083034 | ||
| EMT6/AR1.0 | Function assay | 1 hr | Inhibition of mouse Pgp in EMT6/AR1.0 cells after 1 hr by daunorubicin accumulation assay, IC50=0.064μM | 18083034 | ||
| MDCK | Function assay | 30 mins | Inhibition of P-glycoprotein (unknown origin) expressed in MDCK cells assessed as reduction of calcein-AM transport after 30 mins by fluorescence assay, EC50=0.044μM | 24607999 | ||
| MDCK | Function assay | 30 mins | Activity at MDR1 (unknown origin) expressed in MDCK cells using calcein AM as substrate incubated for 30 mins prior to substrate addition measured after 30 mins by fluorometric analysis, EC50=0.044μM | 23374872 | ||
| NCI-ADR-RES | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 expressed in NCI-ADR-RES cells assessed as potentiation of cytotoxicity by measuring IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 3714.80 +/- 383.58 nM), IC50=0.01851μM | 27504669 | |
| KBV1 | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 expressed in KBV1 cells assessed as potentiation of cytotoxicity by measuring IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 277.68 +/- 56.61 nM), IC50=0.00066μM | 27504669 | |
| HEK293 | Function assay | 1000 nM | 72 hrs | Potentiation of doxorubicin-induced cytotoxicity against HEK293 cells assessed as doxorubicin IC50 at 1000 nM after 72 hrs by CCK8 assay (Rvb = 5.28 +/- 0.74 nM), IC50=0.00495μM | 27504669 | |
| K562/A02 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human K562/A02 cells after 48 hrs by MTT assay, IC50=27.19μM | 28645831 | ||
| SW620 | Cytotoxicity assay | 48 hrs | Cytotoxicity against human SW620 cells assessed as cell viability after 48 hrs by MTT assay, IC50=25μM | 26197160 | ||
| K562/A02 | Function assay | 48 hrs | Inhibition of ABCB1 in human K562/A02 cells assessed as potentiation of adriamycin-induced cytotoxicity by measuring ADR IC50 treated for 48 hrs followed by compound washout measured after 24 hrs by MTT assay (Rvb = 51.34 +/- 5.1 uM), IC50=14.39μM | 28645831 | ||
| MDCK | Function assay | Inhibition of BCRP expressed in MDCK cells by pheophorbide A assay, IC50=0.85μM | 19932960 | |||
| MCF7 MX | Function assay | Inhibition of BCRP expressed in MCF7 MX cells by Hoechst 33342 staining, IC50=0.68μM | 19932960 | |||
| NCI-ADR-RES | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 expressed in NCI-ADR-RES cells assessed as potentiation of doxorubicin-induced cytotoxicity by measuring doxorubicin IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 5.54 +/- 0.60 uM), IC50=0.24μM | 27504669 | |
| MDCK | Function assay | Inhibition of MDR1 expressed in MDCK cells using rhodamine 123 staining by flow cytometry, IC50=0.21μM | 21354800 | |||
| CCRF-CEM/VCR1000 | Function assay | 240 secs | Inhibition of P-glycoprotein-mediated daunorubicin efflux from human CCRF-CEM/VCR1000 cells after 240 secs by FACS flow cytometric analysis, IC50=0.03311μM | 22452412 | ||
| HEK293 | Function assay | 1000 nM | 72 hrs | Inhibition of human ABCB1 transfected in HEK293 cells assessed as potentiation of doxorubicin-induced cytotoxicity by measuring doxorubicin IC50 at 1000 nM after 72 hrs by CCK8 assay (Rvb = 504.65 +/- 44.94 nM), IC50=0.02477μM | 27504669 | |
| KB-3-1 | Function assay | 1000 nM | 72 hrs | Potentiation of cytotoxicity against human KB-3-1 cells assessed as IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 0.78 +/- 0.27 nM), IC50=0.00041μM | 27504669 | |
| OVCAR8 | Function assay | 1000 nM | 72 hrs | Potentiation of cytotoxicity against human OVCAR8 cells assessed as IC50 at 1000 nM after 72 hrs by MTT assay (Rvb = 8.53 +/- 1.95 nM), IC50=0.00518μM | 27504669 | |
| MDCK | Function assay | 30 mins | Activity at BCRP (unknown origin) expressed in MDCK cells using rhodamine 123 as substrate incubated for 30 mins prior to substrate addition measured after 30 mins by fluorometric analysis, EC50=0.01μM | 23374872 | ||
| HepG2 | Function assay | 10 uM | 90 mins | Inhibition of P-gp mediated efflux in adriamycin-resistant human HepG2 cells assessed as intracellular rhodamine-123 accumulation at 10 uM incubated in dark condition for 90 mins by flow cytometry relative to control | 27328029 | |
| Click to View More Cell Line Experimental Data | ||||||
Solubility
|
In vitro |
DMSO
: 8 mg/mL
(12.36 mM)
Water : Insoluble Ethanol : Insoluble |
Molarity Calculator
|
In vivo |
|||||
In vivo Formulation Calculator (Clear solution)
Step 1: Enter information below (Recommended: An additional animal making an allowance for loss during the experiment)
Step 2: Enter the in vivo formulation (This is only the calculator, not formulation. Please contact us first if there is no in vivo formulation at the solubility Section.)
Calculation results:
Working concentration: mg/ml;
Method for preparing DMSO master liquid: mg drug pre-dissolved in μL DMSO ( Master liquid concentration mg/mL, Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug. )
Method for preparing in vivo formulation: Take μL DMSO master liquid, next addμL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O, mix and clarify.
Method for preparing in vivo formulation: Take μL DMSO master liquid, next add μL Corn oil, mix and clarify.
Note: 1. Please make sure the liquid is clear before adding the next solvent.
2. Be sure to add the solvent(s) in order. You must ensure that the solution obtained, in the previous addition, is a clear solution before proceeding to add the next solvent. Physical methods such
as vortex, ultrasound or hot water bath can be used to aid dissolving.
Chemical Information, Storage & Stability
| Molecular Weight | 646.73 | Formula | C38H38N4O6 |
Storage (From the date of receipt) | |
|---|---|---|---|---|---|
| CAS No. | 206873-63-4 | Download SDF | Storage of Stock Solutions |
|
|
| Synonyms | XR9576 | Smiles | COC1=C(C=C2CN(CCC2=C1)CCC3=CC=C(C=C3)NC(=O)C4=CC(=C(C=C4NC(=O)C5=CC6=CC=CC=C6N=C5)OC)OC)OC | ||
Mechanism of Action
| Targets/IC50/Ki |
P-gp
(CHrB30 cells) 5.1 nM(Kd)
|
|---|---|
| In vitro |
Tariquidar displays high-affinity binding to P-gp with Bmax of 275 pmol/mg. This compound shows non-competitive interaction with the P-gp substrates. It increases the steady-state accumulation of these cytotoxics in CHrB30 cells to levels observed in non-P-gp-expressing AuxB1 cells with EC50 of 487 nM. This chemical is able to inhibit the vanadate-sensitive ATPase activity of P-gp by 60-70%, with potent IC50 values of 43 nM. It may inhibit other resistance mechanisms at higher concentrations. 1 μM of this compound abrogates ABCG2 (BCRP)-mediated resistance to camptothecins in vitro. It potentiates the cyto-toxicity of several drugs including doxorubicin; complete reversal of resistance is achieved in the presence of 25- 80 nM of this chemical. In MC26, a murine colon carcinoma cell line with intrinsic chemoresistance, the doxorubicin IC50 is fivefold lower in the presence of 0.1 μM of this compound (36 vs 7 nM). In murine mammary carcinoma, human small-cell lung carcinoma and human ovarian carcinoma cell lines with acquired chemotherapeutic resistance (EMT6/AR1.0, H69/LX4 and 2780 AD), the in vitro doxorubicin IC50 is 22-150-fold lower in the presence of 0.1 μM of this chemical. P-gp inhibition persists for 23 h after removal of it from the culture system. It restored the cyto-toxicity of doxorubicin in the National Cancer Institute (NCI)/ADRRES multicellular tumor spheroid model derived from the MCF7WT breast cancer cell line. |
| Kinase Assay |
Steady-state drug accumulation assay
|
|
Cells are incubated in a reaction volume of 1 mL for 60 min at 37 ℃ under 5% CO2 in order to reach steady-state. The effect of the modulators XR9576 on [3H]-ligand accumulation is investigated in the concentration range 10-9 - 10-6 M. This compound is added from a DMSO stock giving a final solvent concentration of 0.2 % (v/v). Following cell harvesting, accumulated drug is measured by liquid scintillation counting and normalized for cell protein content. Plots of amount accumulated as a function of modulator concentration are fitted with the general dose-response equation: Y={(a-b)/(1+(X/c)d)}+bWhere: Y=response; a=initial response; b=final response; c=EC50 concentration; d=slope value; X=drug concentration.
|
|
| In vivo |
Tariquidar (2- 8 mg/kg p.o.) is found to significantly potentiate the antitumor activity of doxorubicin (5 mg/kg, i.v.) against MC26 murine colon carcinoma in vivo. In human carcinoma xenografts, coadministration of this compound (6 -12 mg/kg p.o.) fully restored the antitumor activity against two highly resistant MDR human tumor xenografts (2780AD, H69/LX4) in nude mice. |
References |
|
Applications
| Methods | Biomarkers | Images | PMID |
|---|---|---|---|
| Immunofluorescence | MRP7 |
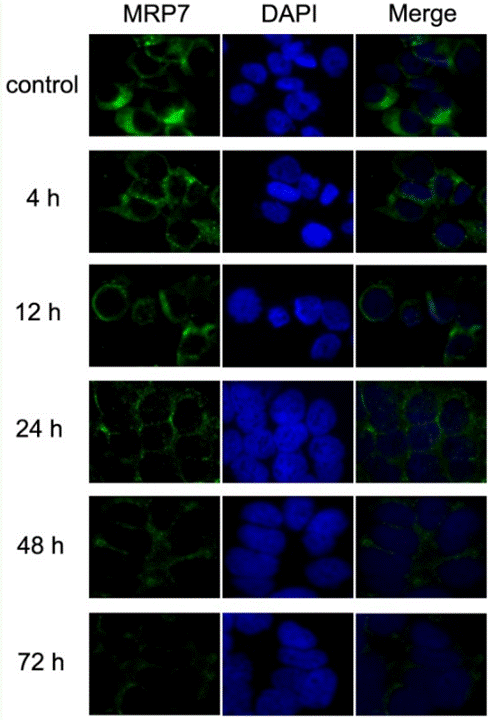
|
23393594 |
Clinical Trial Information
(data from https://clinicaltrials.gov, updated on 2024-05-22)
| NCT Number | Recruitment | Conditions | Sponsor/Collaborators | Start Date | Phases |
|---|---|---|---|---|---|
| NCT01663545 | Completed | Epilepsies Partial |
National Institute of Neurological Disorders and Stroke (NINDS)|National Institutes of Health Clinical Center (CC) |
July 31 2012 | -- |
| NCT01547754 | Terminated | HIV-Associated Cognitive Motor Complex |
National Institute of Mental Health (NIMH)|National Institutes of Health Clinical Center (CC) |
January 9 2012 | -- |
| NCT01386476 | Completed | Drug Resistance |
National Institute of Mental Health (NIMH)|National Institutes of Health Clinical Center (CC) |
June 15 2011 | -- |
| NCT00082368 | Completed | Cancer |
National Cancer Institute (NCI)|National Institutes of Health Clinical Center (CC) |
May 16 2004 | Phase 2 |
Tech Support
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
Frequently Asked Questions
Question 1:
Can you please give me more specific and detailed information of how to dissolve and use it (S8028) for in vivo studies?
Answer:
It in 30% Propylene glycol, 5% Tween 80, 65% D5W at 30mg/ml will be a suspension or emulsion. If you are going to administrate this compound by oral gavage, it is fine. We also have test some vehicles for it for i.p injection, and it is soluble in 5% DMSO+45% PEG 300+ddH2O at 2mg/ml clearly. When preparing the solution, please dissolve it in DMSO clearly first, then add PEG. After they mixed well, then dilute with water.
Signaling Pathway Map
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.