
- Bioactive Compounds
- By Signaling Pathways
- PI3K/Akt/mTOR
- Epigenetics
- Methylation
- Immunology & Inflammation
- Protein Tyrosine Kinase
- Angiogenesis
- Apoptosis
- Autophagy
- ER stress & UPR
- JAK/STAT
- MAPK
- Cytoskeletal Signaling
- Cell Cycle
- TGF-beta/Smad
- Compound Libraries
- Antibodies
- Bioreagents
- qPCR
- 2x SYBR Green qPCR Master Mix
- 2x SYBR Green qPCR Master Mix(Low ROX)
- 2x SYBR Green qPCR Master Mix(High ROX)
- Protein Assay
- Protein A/G Magnetic Beads for IP
- Anti-Flag magnetic beads
- Anti-Flag Affinity Gel
- Anti-Myc magnetic beads
- Anti-HA magnetic beads
- Poly FLAG Peptide lyophilized powder
- Protease Inhibitor Cocktail
- Protease Inhibitor Cocktail (EDTA-Free, 100X in DMSO)
- Phosphatase Inhibitor Cocktail (2 Tubes, 100X)
- Cell Biology
- Cell Counting Kit-8 (CCK-8)
- Animal Experiment
- Mouse Direct PCR Kit (For Genotyping)
- New Products
- Contact Us
-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
-
 Other Countries
Other Countries
NF-κB
NF-κB Products
- All (139)
- NF-κB Inhibitors (123)
- NF-κB Antibodie (1)
- NF-κB Activators (3)
- NF-κB Modulators (8)
- New NF-κB Products
| Catalog No. | Product Name | Information | Product Use Citations | Product Validations |
|---|---|---|---|---|
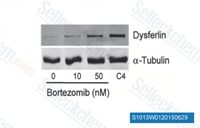
| S1013 | Bortezomib | Bortezomib is a potent 20S proteasome inhibitor with Ki of 0.6 nM. It exhibits favorable selectivity towards tumor cells over normal cells. Bortezomib (PS-341) inhibits NF-κB and induces ERK phosphorylation to suppress cathepsin B and inhibit the catalytic process of autophagy in ovarian cancer and other solid tumors. |
|
|
| S1623 | Acetylcysteine (N-acetylcysteine) | Acetylcysteine (N-acetyl-l-cysteine, NAC,N-acetylcysteine) is a ROS(reactive oxygen species) inhibitor that antagonizes the activity of proteasome inhibitors. It is also a tumor necrosis factor production inhibitor. Acetylcysteine(N-acetyl-l-cysteine) suppresses TNF-induced NF-κB activation through inhibition of IκB kinases. Acetylcysteine(N-acetyl-l-cysteine) induces apoptosis via the mitochondria-dependent pathway. Acetylcysteine(N-acetyl-l-cysteine) inhibits ferroptosis and virus replication.Solutions are unstable and should be fresh-prepared. |

|
|
| S7351 | JSH-23 | JSH-23 is an inhibitor of NF-κB transcriptional activity with IC50 of 7.1 μM in RAW 264.7 cell line. |

|
|
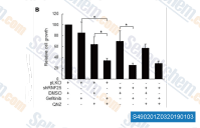
| S4902 | QNZ (EVP4593) | QNZ (EVP4593) shows potent inhibitory activity toward both NF-κB activation and TNF-α production with IC50 of 11 nM and 7 nM in Jurkat T cells, respectively. |
|
|
| S3633 | PDTC (Pyrrolidinedithiocarbamate ammonium) | PDTC (Pyrrolidinedithiocarbamate ammonium) is a potent nuclear factor-κB (NF-κB) inhibitor that inhibits IκB phosphorylation, blocks NF-κB translocation to the nucleus and reduces the expression of downstream cytokines. | ||
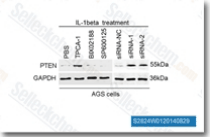
| S2824 | TPCA-1 | TPCA-1 (GW683965) is an inhibitor of IKK-2 with IC50 of 17.9 nM in a cell-free assay, inhibits NF-κB pathway, exhibits 22-fold selectivity over IKK-1. TPCA-1 is also an inhibitor of STAT3 and enhances apoptosis. |
|
|
| S7273 | SC75741 | SC75741 is a potent NF-κB inhibitor with EC50 of 200 nM. SC75741 efficiently blocks influenza virus propagation. |

|
|
| S7414 | Caffeic Acid Phenethyl Ester | Caffeic acid phenethyl ester (CAPE, Phenylethyl Caffeate) is a potent and specific inhibitor of NF-κB activation, and also displays antioxidant, immunomodulatory and antiinflammatory activities. |

|
|
| S1848 | Curcumin | Curcumin (Diferuloylmethane, Natural Yellow 3, Turmeric yellow) is the principal curcuminoid of the popular Indian spice turmeric, which is a member of the ginger family (Zingiberaceae). It is an inhibitor of p300 histone acetylatransferase(IC50~25 μM)and Histone deacetylase (HDAC); activates Nrf2 pathway and supresses the activation of NF-κB. Curcumin induces mitophagy, autophagy, apoptosis, and cell cycle arrest with antitumor activity. Curcumin reduces renal damage associated with rhabdomyolysis by decreasing ferroptosis-mediated cell death. Curcumin exhibits anti-infective properties against various human pathogens like the influenza virus, hepatitis C virus, HIV and so on. |

|
|
| S8341 | TAK-243 (MLN7243) | TAK-243 (MLN7243) is a potent, mechanism-based small-molecule inhibitor of the ubiquitin activating enzyme (UAE) with an IC50 of 1 ± 0.2 nM in the UBCH10 E2 thioester assay. It has minimal inhibitory activity in a panel of kinase and receptor assays, as well as on human carbonic anhydrase type I and type II. TAK-243 (MLN7243) induces ER stress, abrogates NFκB pathway activation and promotes apoptosis. | ||
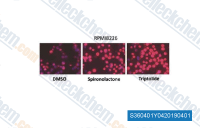
| S3604 | Triptolide | Triptolide is a diterpene triepoxide, immunosuppresive agent extracted from the Chinese herb Tripterygium wilfordii. It functions as a NF-κB inhibitor with dual actions by disruption of p65/CBP interaction and by reduction of p65 protein. Triptolide (PG490) abrogates the transactivation function of heat shock transcription factor 1 (HSF1). Triptolide inhibits MDM2 and induces apoptosis through a p53-independent pathway. |
|
|
| S8279 | Shikonin | Shikonin, a potent and specific Pyruvate kinase M2 (PKM2) inhibitor, is a major component of zicao (purple gromwell, the dried root of Lithospermum erythrorhizon), a Chinese herbal medicine with various biological activities. It is also an inhibitor of TMEM16A chloride channel activity using cell-based fluorescent-quenching assay. Shikonin exerts an anti-inflammatory effect by inhibiting tumor necrosis factor-α (TNF-α) and prevents activation of nuclear factor-κB (NF-κB) pathway via proteasome inhibition. | ||
| S8078 | Bardoxolone Methyl | Bardoxolone Methyl (RTA 402, TP-155, NSC 713200, CDDO Methyl Ester, CDDO-Me) is an IKK inhibitor, showing potent proapoptotic and anti-inflammatory activities; Also a potent Nrf2 activator and nuclear factor-κB (NF-κB) inhibitor. Bardoxolone Methyl abrogates ferroptosis. Bardoxolone methyl induces apoptosis and autophagy in cancer cells. |

|
|
| S2290 | DHA (Dihydroartemisinin) | DHA (Dihydroartemisinin) is a semi-synthetic derivative of artemisinin and isolated from the traditional Chinese herb Artemisia annua. Dihydroartemisinin induces autophagy and apoptosis by suppressing NF-κB activation. |

|
|
| S2341 | (-)-Parthenolide | (-)-Parthenolide, an inhibitor of the Nuclear Factor-κB Pathway, specifically depletes HDAC1 protein without affecting other class I/II HDACs; Also promotes the ubiquitination of MDM2 and activates p53 cellular functions. |
-Parthenolide-S234101W0220161110.gif)
|
|
| S1576 | Sulfasalazine | Sulfasalazine is a sulfa derivative of mesalazine, used as an anti-inflammatory agent to treat bowel disease and rheumatoid arthritis. Sulfasalazine is a potent and specific inhibitor of nuclear factor kappa B (NF-κB), TGF-β and COX-2. Sulfasalazine induces ferroptosis, apoptosis and autophagy. | ||
| S5771 | Sulforaphane | Sulforaphane is a naturally occurring isothiocyanate derived from the consumption of cruciferous vegetables, such as broccoli, cabbage, and kale. It is an inducer of Nrf2. Sulforaphane is also an inhibitor of histone deacetylase (HDAC) and NF-κB. Sulforaphane increases heme oxygenase-1 (HO-1) and reduces the levels of reactive oxygen species (ROS). Sulforaphane induces cell cycle arrest and apoptosis., | ||
| S3603 | Betulinic acid | Betulinic acid (ALS-357, Lupatic acid, Betulic acid), a pentacyclic triterpenoid from Syzigium claviflorum, is a inhibitor of HIV-1 with EC50 of 1.4 μ M. It's reported that Betulinic acid acts as a new activator of NF-κB.Phase 1/2. | ||
| S2261 | Andrographolide | Andrographolide is a labdane diterpenoid that is the main bioactive component of the medicinal plant Andrographis paniculata. | ||
| S2382 | Evodiamine | Evodiamine (Isoevodiamine), an alkaloid extract from the fruit of Evodiae Fructus exhibits antitumor activities against the human tumor cells. It is shown to inhibit NF-κB activation through suppression of IkB kinase activity. |
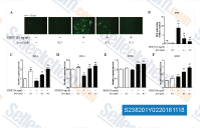
|
|
| S8587 | Withaferin A (WFA) | Withaferin A (WFA) potently inhibits NF-κB activation by preventing the tumor necrosis factor-induced activation of IκB kinase β via a thioalkylation-sensitive redox mechanism. Withaferin A binds to the intermediate filament (IF) protein, vimentin with antitumor and antiangiogenesis activity. Withaferin A is a steroidal lactone isolated from Withania somnifera. | ||
| S5804 | N-Acetylcysteine amide | N-acetylcysteine amide is a membrane penetrating antioxidant with anti-inflamatory activity through regulation of activation of NF-κB and HIF-1α as well as modulation of ROS. | ||
| S8483 | CBL0137 HCl | CBL0137 (CBLC137, Curaxin 137) HCl activates p53 and inhibits NF-κB with EC50s of 0.37 μM and 0.47 μM in the cell-based p53 and NF-kB reporter assays, respectively. It also inhibits histone chaperone FACT (facilitates chromatin transcription complex). | ||
| S0507 | CBL0137 | CBL0137 (Curaxin-137) is an inhibitor of the histone chaperone FACT (facilitates chromatin transcription) that simultaneously suppresses NF-κB and activates p53 with EC50 of 0.47 μM and 0.37 μM, respectively. | ||
| S2321 | Magnolol | Magnolol (NSC 293099) is a bioactive lignin found in the bark of the Houpu magnolia (Magnolia officinalis) which shows antifungal properties. It can block TNF-α-induced NF-κB activation. |

|
|
| S7672 | Omaveloxolone (RTA-408) | Omaveloxolone (RTA-408) is a synthetic triterpenoid that activates the cytoprotective transcription factor Nrf2 and inhibits NF-κB signaling. Phase 2. | ||
| S9141 | Berbamine | Berbamine (BA), a traditional Chinese medicines extracted from Berberis amurensis (xiaoboan), is a novel inhibitor of bcr/abl fusion gene with potent anti-leukemia activity and also an inhibitor of NF-κB. Berbamine (BA) induces apoptosis in human myeloma cells and inhibits the growth of cancer cells by targeting Ca²⁺/calmodulin-dependent protein kinase II (CaMKII). | ||
| S5453 | Hyperoside | Hyperoside (Hyperin, Quercetin 3-galactoside), a naturally occuring flavonoid compound, exerts multiple bioactivities, including myocardial protection, anti-redox, and anti-inflammatory activities.Hyperoside can inhibit activation of the NF-κB signaling pathway. | ||
| S5144 | Neferine | Neferine ((R)-1,2-Dimethoxyaporphine), a natural component of Nelumbo nucifera, has antitumor efficiency. Neferine induces apoptosis in renal cancer cells. Neferine prevents autophagy through activation of Akt/mTOR pathway and Nrf2 in muscle cells. Neferine strongly inhibits NF-κB activation. Neferine possesses a number of therapeutic effects such as anti-diabetic, anti-aging, anti-microbial, anti-thrombotic, anti-arrhythmic, anti-inflammatory and even anti-HIV. | ||
| S3901 | Astragaloside IV | Astragaloside IV (AST-IV, AS-IV) is a bioactive saponin first isolated from the dried plant roots of the genus Astragalus, which is used in traditional Chinese medicine. It has various effect on the cardiovascular, immune, digestive, and nervous systems. AS-IV suppresses activation of p-Akt, p-mTOR, p-NF-κB and p-Erk1/2. | ||
| S6671 | SN50 | SN50 (NF-κB SN50), a cell-permeable NF-κB inhibitory peptide, is composed of the signal peptide of Kaposi fibroblast growth factor.SN50 inhibits the activation of NF-κB and attenuates ventilator-induced lung injury. | ||
| S7445 | APX-3330 | APX-3330 (E3330) is a potent and selective APE1(Ref-1) inhibitor, which suppressed NF-kappa B DNA-binding activity. | ||
| S3931 | Ginsenoside Rd | Ginsenoside Rd (Panaxoside Rd, Sanchinoside Rd), a minor ginseng saponin, has several pharmacological activities such as immunosuppressive activity, anti-inflammatory activity, immunological adjuvant, anti-cancer activity and wound-healing activity. Ginsenoside Rd inhibits TNFα-induced NF-κB transcriptional activity with an IC50 of 12.05±0.82 μM in HepG2 cells. Ginsenoside Rd inhibits expression of COX-2 and iNOS mRNA. Ginsenoside Rd also inhibits Ca2+ influx. Ginsenoside Rd inhibits CYP2D6, CYP1A2, CYP3A4, and CYP2C9, with IC50s of 58.0±4.5 μM, 78.4±5.3 μM, 81.7±2.6 μM, and 85.1±9.1 μM, respectively. | ||
| S3771 | Stachydrine | Stachydrine (Proline betaine, L-stachydrine, Methyl hygrate betaine) is a quaternary ammonium derivative of proline that occurs widely in Medicago species. It is an osmoprotective compound found in urine. Stachydrine is a major constituent of Chinese herb leonurus heterophyllus sweet used to promote blood circulation and dispel blood stasis. Stachydrine can inhibit the NF-κB signal pathway. | ||
| S3874 | Curcumenol | Curcumenol, a sesquiterpene isolated from Curcuma zedoaria, is known to possess a variety of health and medicinal values which includes neuroprotection, anti-inflammatory, anti-tumor and hepatoprotective activities. It inhibits NF-κB activation by suppressing the nuclear translocation of the NF-κB p65 subunit and blocking IκBα phosphorylation and degradation. | ||
| S3924 | Ginsenoside Rb1 | Ginsenoside Rb1 (Gypenoside Ⅲ) is a protopanaxadiol that has diverse in vitro and in vivo effects, including neuroprotective, anti-inflammatory, and anti-obesity actions. Ginsenoside Rb1, a main constituent of the root of Panax ginseng, inhibits Na+, K+-ATPase activity with IC50 of 6.3±1.0 μM. Ginsenoside also inhibits IRAK-1 activation and phosphorylation of NF-κB p65. Ginsenoside Rb1 reduces the expressions of TLR3, TLR4 and TRAF-6, and down-regulates the levels of TNF-α, IFN-β and iNOS. | ||
| S4073 | Sodium 4-Aminosalicylate | Sodium 4-Aminosalicylate is an antibiotic used to treat tuberculosis via NF-κB inhibition and free radical scavenging. |
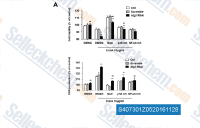
|
|
| E0796 | NF-κB-IN-1 | NF-κB-IN-1 is a potent NF-κB signaling pathway inhibitor, which acts by directly inhibiting inhibitory κB kinase (IKK). | ||
| S9193 | Aristolochic acid A | Aristolochic acid A (Aristolochic Acid I, Aristolochin, Aristolochine, TR 1736) is a carcinogenic, mutagenic, and nephrotoxic agent extracted from the flowering plant family Aristolochiaceae.Aristolochic acid A significantly reduces both activator protein 1 (AP-1) and nuclear factor-κB (NF-κB) activities. | ||
| S7428 | Rocaglamide | Rocaglamide (Roc-A), isolated from Aglaia species, is a potent inhibitor of heat shock factor 1 (HSF1) activation with IC50 of ~50 nM for HSF1. Rocaglamide inhibits the function of the translation initiation factor eIF4A. Rocaglamide also inhibits NF-κB activity. Rocaglamide exhibits anti-tumor activity. | ||
| S2416 | Chondroitin sulfate | Chondroitin sulfate (CS) is a major component of the extracellular matrix (ECM) of many connective tissues, including cartilage, bone, skin, ligaments and tendons. It reduces the IL-1β-induced nuclear factor-kB (NF-κB) translocation in chondrocytes in vitro. | ||
| S9170 | Engeletin | Engeletin (Dihydrokaempferol 3-rhamnoside), a bioactive flavonoid, has multiple biological activities such as anti-diabetic and anti-inflammatory effects. engeletin againsts LPS-stimulated endometritis in mice via negative regulation of pro-inflammatory mediators via the TLR4-regulated NF-κB pathway. | ||
| S3923 | Ginsenoside Rg1 | Ginsenoside Rg1 (Ginsenoside A2, Panaxoside A, Panaxoside Rg1, Sanchinoside C1, Sanchinoside Rg1), one of the major active components of ginseng, is identified as a protopanaxatriol-type and has pharmacological actions such as neuroprotective and anti-tumor effects on various cancer types. Ginsenoside Rg1 reduces cerebral Aβ levels and NF-κB nuclear translocation. | ||
| S2979 | CU-T12-9 | CU-T12-9 is a specific agonist of Toll-like receptor (TLR) 1/2 with EC50 of 52.9 nM in HEK-Blue hTLR2 SEAP assay. CU-T12-9 activates both the innate and the adaptive immune systems. CU-T12-9 signals through NF-κB and invokes an elevation of the downstream effectors TNF-α, IL-10, and iNOS. | ||
| S3137 | Sodium salicylate | Sodium salicylate(2-Hydroxybenzoic acid Sodium salt,Salicylic acid Sodium salt) is used in medicine as an analgesic and antipyretic. | ||
| S4798 | Benfotiamine | Benfotiamine (S-Benzoylthiamine O-monophosphate) is a synthetic S-acyl derivative of thiamine (vitamin B1) and has been investigated for the treatment and prevention of Diabetic Nephropathy and Diabetes Mellitus, Type 2. Benfotiamine suppresses oxidative stress-induced NF-κB activation and prevents the bacterial endotoxin-induced inflammation. | ||
| A2020 | Denosumab (anti-RANK ligand) | Denosumab (anti-RANK ligand) (AMG-162) is a fully human monoclonal antibody that selectively inhibits primate RANKL. Denosumab inhibits the ability of human RANKL (143–317) to stimulate the formation of osteoclasts derived from murine RAW 264.7 cells with an IC50 value of 1.64 nM. | ||
| S5640 | Ethyl caffeate | Ethyl caffeate, a naturally occurring compound found in Bidens pilosa, suppresses NF-kappaB activation and its downstream inflammatory mediators, iNOS, COX-2 and PGE2 in vitro. | ||
| S9295 | Dauricine | Dauricine, a plant metabolite isolated from the Asian vine Menispermum dauricum, plays a variety of biological roles in the human body, from inhibiting cancer cell growth to blocking cardiac transmembrane Na+, K+, and Ca2+ ion currents.Dauricine induces apoptosis, inhibits proliferation and invasion through inhibiting NF‐κB signaling pathway in colon cancer cells. | ||
| S3770 | Sodium Aescinate | Sodium Aescinate (SA, Escin Sodium Salt) is a widely-applied triterpene saponin product derived from horse chestnut seeds, possessing vasoactive and organ-protective activities with oral or injection administration in the clinic. Sodium aescinate is a triterpene saponin derived from Aesculus hippocastanum seeds, with anti-inflammatory and antioxidant activities. Sodium aescinate inhibits hepatocellular carcinoma growth by targeting CARMA3/NF-κB pathway. | ||
| S9374 | 2',5'-Dihydroxyacetophenone | 2',5'-Dihydroxyacetophenone (DHAP, 2-Acetylhydroquinone, Quinacetophenone) is a mixture of dihydroxyacetophenone isomers is used in food flavouring. 2',5'-Dihydroxyacetophenone significantly inhibits NO production via the suppression of iNOS expression. 2',5'-Dihydroxyacetophenone significantly decreases levels of the pro-inflammatory cytokines TNF-α and IL-6 by blocking the ERK1/2 and NF-κB signaling pathways. | ||
| S3871 | Muscone | Muscone (3-Methylcyclopentadecanone, Methylexaltone), a flavouring ingredient, is an organic compound that is the primary contributor to the odor of musk and also a potent anti-inflammatory agent. Muscone significantly downregulats the levels of LPS-induced inflammatory cytokines and inhibits NF-κB and NLRP3 inflammasome activation in BMDMs. Muscone remarkably decreases the levels of inflammatory cytokines (IL-1β, TNF-α and IL-6). | ||
| S7637 | DTP3 | DTP3 is a selective GADD45β/MKK7 (growth arrest and DNA-damage-inducible β/mitogen-activated protein kinase kinase 7) inhibitor and is able to restore MKK7/JNK activation. DTP3 inhibits cancer-selective NF-κB survival pathway. | ||
| S4763 | 4-Hydroxychalcone | 4-Hydroxychalcone (P-Cinnamoylphenol) is a chalcone metabolite with diverse biological activities. It inhibits TNFα-induced NF-κB pathway activation in a dose-dependent manner and also activates BMP signaling. | ||
| S3808 | Mangiferin | Mangiferin (Alpizarin, Chinomin, Hedysarid) is a bioactive compound that demonstrates many health perspectives and has been used to prepare medicinal and food supplements. Mangiferin is a Nrf2 activator. Mangiferin suppresses nuclear translocation of the NF-κB subunits p65 and p50. | ||
| S3810 | Scutellarin | Scutellarin (Breviscapine, Breviscapin, Scutellarein-7-glucuronide), the major active principal flavonoids extracted from the Chinese herbal medicines Scutellaria baicalensis and Erigeron breviscapus (Vant.) Hand-Mazz, has many pharmacological effects, such as antioxidant, antitumor, antiviral, and antiinflammatory activities. Scutellarin can down-regulates the STAT3/Girdin/Akt signaling in HCC cells, and inhibits RANKL-mediated MAPK and NF-κB signaling pathway in osteoclasts. | ||
| S3607 | Sarsasapogenin | Sarsasapogenin (SAR, Parigenin) is a steroidal sapogenin. It can provoke the generation of reactive oxygen species and activate unfolded protein response (UPR) signaling pathways. SAR potently inhibits NF-κB and MAPK activation, as well as IRAK1, TAK1, and IκBα phosphorylation in LPS-stimulated macrophages. | ||
| S3811 | Ginsenoside Re | Ginsenoside Re (Ginsenoside B2, Panaxoside Re, Sanchinoside Re, Chikusetsusaponin Ivc), an extract from Panax notoginseng, is a major ginsenoside in ginseng and belongs to 20(S)-protopanaxatriol group. It has diverse in vitro and in vivo effects, including vasorelaxant, antioxidant, antihyperlipidemic, and angiogenic actions. Ginsenoside Re decreases the β-amyloid protein (Aβ). Ginsenoside Re plays a role in antiinflammation through inhibition of JNK and NF-κB. | ||
| S9502 | Madecassic acid | Madecassic acid (Brahmic acid), a natural triterpene first isolated from C. asiatica, has diverse anti-inflammatory and anti-diabetic effects, blocking NF-κB activation in macrophages and causing by iNOS, COX-2, TNF-alpha, IL-1beta and IL-6 inhibition. | ||
| S0454 | Adjudin | Adjudin (AF-2364), a potent male contraceptive, is a potent blocker of Cl⁻ channels. Adjudin exhibits anti-inflammatory properties by suppression of NF-κB p65 nuclear translocation and DNA binding activity as well as ERK MAPK phosphorylation. | ||
| S3150 | Articaine HCl | Articaine (Ultracaine) is a dental local anesthetic which contains an additional ester group that is metabolized by estearases in blood and tissue. | ||
| S3939 | 4'-Methoxyresveratrol | 4-Methoxyresveratrol is a stibenoid found in the Chinese herb Gnetum cleistostachyum. 4'-Methoxyresveratrol alleviates AGE-induced inflammation through suppressing RAGE-mediated MAPK/NF-κB signaling pathway and NLRP3 inflammasome activation. | ||
| S4611 | TCEP Hydrochloride | TCEP (Tris(2-carboxyethyl)phosphine) hydrochloride, a non-thiol reducing agent, promotes NF-κB-DNA binding in a dose-related manner.Solutions are best fresh-prepared. | ||
| E0212 | Cornuside | Cornuside is a bisiridoid glucoside compound isolated from the fruit of Cornus officinalis SIEB. et ZUCC. Cornuside suppresses lipopolysaccharide-induced inflammatory mediators by inhibiting NF-κB activation in RAW 264.7 macrophages. Cornuside attenuates apoptosis in rat cortical neurons. |
||
| E3034 | Cynanchi Atrati Extract | Cynanchi Atrati Extract is extracted from Cynanchi atrati Radix, has been traditionally used as an anti-inflammatory agent, and has a potential to inhibit IKK-mediated NF-κB activation, which is valuable for modulating inflammatory or cancerous conditions. | ||
| S0002 | IAXO-102 | IAXO-102 is an antagonist of TLR4 targeting both MD-2 and CD14 co-receptors. IAXO-102 inhibits MAPK and p65 NF-κB phosphorylation and downregulates the expression of TLR4 dependent proinflammatory proteins. IAXO-102 inhibits experimental abdominal aortic aneurysm (AAA) development. | ||
| S6899 | Licochalcone D | Licochalcone D (Lico D, LCD, LD), a flavonoid isolated from a Chinese medicinal plant Glycyrrhiza inflata, has antioxidant, anti-inflammatory and anti-cancer properties. Licochalcone D inhibit phosphorylation of NF-κB p65 in LPS signaling pathway. Licochalcone D inhibits JAK2, EGFR and Met (c-Met) activities and induces ROS-dependent apoptosis. Licochalcone D also induces caspases activation and poly (ADP-ribose) polymerase (PARP) cleavage. | ||
| S0911 | Hypaphorine | Hypaphorine which is abundantly found in vaccaria semen, counteracts inflammation via inhibition of ERK or/and NFκB signaling pathways. | ||
| S3223 | L-Quebrachitol | L-Quebrachitol (L-QCT), a natural product isolated from many plants, promotes proliferation and cell DNA synthesis. L-Quebrachitol upregulates bone morphogenetic protein-2 (BMP-2) and runt-related transcription factor-2 (Runx2) and regulatory genes associated with mitogen-activated protein kinase (MAPK) and Wnt/β-catenin signaling pathway, while down-regulating the receptor activator of the nuclear factor-κB(NF-κB) ligand (RANKL) mRNA level. | ||
| S3998 | (+)-α-Lipoic acid | (+)-α-Lipoic acid ((R)-(+)-α-Lipoic acid, α-Lipoic acid, Alpha-Lipoic acid), a physiological form of thioctic acid, is a strong antioxidant that relieves diabetic neuropathic symptoms. It shows superior antioxidative effects to its racemate. R(+)-α-lipoic acid is an essential cofactor of mitochondrial enzyme complexes. R(+)-α-lipoic acid inhibits NF-κB-dependent HIV-1 LTR activation. | ||
| S0915 | Morusin | Morusin (Mulberrochromene) is an inhibitor of NF-κB and STAT3. Morusin is a prenylated flavonoid isolated from Marsdenia australis with antitumor, antioxidant, and anti-bacteria properties. | ||
| S9208 | Ginsenoside Rb3 | Ginsenoside Rb3, extracted from the plant Panax ginseng, plays important roles in cardiovascular diseases, including myocardial ischemia-reperfusion (I/R) injury. Ginsenoside Rb3 (0.1-10 μM) is tested for inhibition of tumor necrosis factor-α (TNF)-induced nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) luciferase reporter activity using a human kidney 293T cell-based assay. Ginsenoside Rb3 shows the significant activity with an IC50 of 8.2 μM. Ginsenoside Rb3 also inhibits the induction of cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) messenger Ribonucleic acid (mRNA) in a dose-dependent manner after HepG2 cells have been treated with TNF-α (10 ng/mL). | ||
| S0923 | Isoliquiritin apioside | Isoliquiritin apioside (ISLA, ILA), a component isolated from Glycyrrhizae radix rhizome (GR), significantly decreases PMA-induced increases in matrix metalloproteinase (MMP) activities and suppresses PMA-induced activation of mitogen-activated protein kinase (MAPK) and NF-κB. Isoliquiritin apioside possesses anti-metastatic and anti-angiogenic abilities in malignant cancer cells and ECs, with no cytotoxicity. | ||
| S8961 | Alobresib (GS-5829) | Alobresib (GS-5829) is a novel BET inhibitor that represents a highly effective therapeutics agent against recurrent/chemotherapy-resistant USC-overexpressing c-Myc. Alobresib (GS-5829) inhibits CLL cell proliferation and induces leukemia cell apoptosis through deregulation of key signaling pathways, such as BLK, AKT, ERK1/2, and MYC. Alobresib (GS-5829) also inhibits NF-κB signaling. | ||
| E4443New | S-Nitrosoglutathione | S-nitrosoglutathione (GSNO, RVC-588, S-Nitroso-L-glutathione,Nitrosoglutathione) is an amino acid and nitric oxide donor that, under physiological conditions, spontaneously releases nitric oxide. S-nitrosoglutathione (GSNO) selectively inhibits platelet and NF-κB activation. It induces apoptosis in T cells in low concentrations (< 0.6 mM) while protecting T cells from apoptosis in higher concentrations (1-2 mM). | ||
| S0931 | Jaceosidin | Jaceosidin, a flavonoid isolated from Artemisia vestita, possesses anti-tumor and anti-proliferative activities in many cancer cells. Jaceosidin induces apoptosis, activates Bax and down-regulates Mcl-1 and c-FLIP expression. Jaceosidin inhibits COX-2 expression and NF-κB activation. | ||
| S3243 | Zeaxanthin | Zeaxanthin, the carotenoid alcohol participates in the xanthophyll cycle, activates the extrinsic apoptosis pathway which induces apoptosis on uveal melanoma cells with IC50 value 40.8 µM. | ||
| S3231 | UCB-9260 | UCB-9260 is an orally active inhibitor that inhibits TNF signalling by stabilising an asymmetric form of the trimer. UCB-9260 binds TNF with Kd of 13 nM. UCB-9260 also inhibits NF-κB with IC50 of 202 nM after TNF stimulation. | ||
| E3566 | Largeleaf Gentian Root Extract | Largeleaf Gentian Root Extract is extracted from the root of Gentiana macrophylla, which can modulate the CD147/p38/NF-κB pathway. | ||
| S8989 | Xanthatin | Xanthatin is a sesquiterpene lactone isolated from Xanthium strumarium leaves, which can inhibit the nuclear factor kappa-B (NF-κB), mitogen-activated protein kinase (MAPK) and signal transducer and activator of transcription (STATs) signaling pathways. | ||
| S0949 | Cucurbitacin IIb | Cucurbitacin IIb (CuIIb, Dihydrocucurbitacin F, 25-deacetyl hemslecin A) inhibits phosphorylation of STAT3, JNK and Erk1/2, enhances the phosphorylation of IκB and NF-κB, blocks nuclear translocation of NF-κB and decreases mRNA levels of IκBα and TNF-α. Cucurbitacin IIb exhibits anti-inflammatory activity and induces apoptosis. Cucurbitacin IIb is isolated from Hemsleya amabilis. | ||
| S3816 | Dehydroevodiamine | Dehydroevodiamine (DHED), a constituent of Evodia rutaecarpa, has various biological effects such as hypotensive, negative chronotropic, ion channel depressant, inhibition of nitric oxide production and cerebral blood flow enhancing activities. Dehydroevodiamine inhibits LPS-induced iNOS, COX-2, prostaglandin E2 (PGE2) and nuclear factor-kappa B (NF-κB) expression in murine macrophage cells. | ||
| S9244 | 8-O-acetyl shanzhiside methyl ester | 8-O-acetyl shanzhiside methyl ester (Barlerin, ND01), isolated from the leaves of Lamiophlomis rotata Kudo, promotes angiogenesis, which leads to the improvement of functional outcome after stroke.8-O-Acetyl shanzhiside methyl ester can inhibts NF-κB. | ||
| S3384 | (E/Z)-IT-603 | (E/Z)-IT-603 is a mixture of E-IT-603 and Z-IT-603. IT-603 is a NF-κB family member c-Rel inhibitor with an IC50 of 3 μM in electrophoretic mobility shift assay (EMSA). | ||
| P1238New | SN 52 | SN52, a cell-permeable peptide containing the p50 NLS, is a selective inhibitor of NF-κB pathway. It selectively and completely blocks the ionizing radiation (IR)-induced nuclear import of p52 and RelB. It also enhances the radiosensitivity of prostate cancer cells. | ||
| E3652 | Rehmanniae radix praeparata Extract | Rehmanniae radix praeparata Extract is extracted from Rehmanniae radix praeparata, which can improve diabetes through AMPK-mediated NF-κB/NLRP3 signaling pathway. | ||
| E3654 | Althaea rosea Extract(root) | Althaea rosea Extract(root) is extracted from the root of Althaea rosea Extract, which can inhibit NF-κB. | ||
| E1575New | EN450 | EN450 is a cysteine-reactive covalent molecular glue degrader targeting NF-κB. It interacts with allosteric C111 in the E2 ubiquitin ligase UBE2D that induces the ternary complex formation between UBE2D and NFKB1. It exhibits anti-proliferative effects through a Cullin E3 ligase and proteasome-dependent mechanism. | ||
| S0781 | IQ 3 | IQ 3 is a specific c-Jun N-terminal kinase (JNK) inhibitor with Kd of 66 nM, 240 nM and 290 nM for JNK3, JNK1 and JNK2, respectively. IQ 3 inhibits LPS-induced NF-κB/AP1 transcriptional activity in THP1-Blue cells with IC50 of 1.4 μM. IQ 3 also inhibits TNF-α and IL-6 production in vitro. | ||
| E0022 | Ophiopogonin D | Ophiopogonin D (OJV-VI, Deacetylophiopogonin C) is a steroidal glycoside isolated from Chinese herb Radix ophiopogonis and shows anti-tumor property. Ophiopogonin D could suppress TGF-β1-mediated metastatic behavior of MDA-MB-231 cells by regulating ITGB1/FAK/Src/AKT/β-catenin/MMP-9 signaling axis. Ophiopogonin D attenuates PM2.5-induced inflammation via suppressing the AMPK/NF-κB pathway in mouse pulmonary epithelial cells. | ||
| S4937 | 4'-Hydroxychalcone | 4'-Hydroxychalcone (P-Cinnamoylphenol), found in herbs and spices and tea, is a member of the class of compounds known as retrochalcones. It has diverse biological activities, inhibiting TNFα-induced NF-κB pathway activation in a dose-dependent manner and activating BMP signaling. | ||
| S5343 | Vanillic acid | Vanillic acid (4-hydroxy-3-methoxybenzoic acid) is a flavoring agent which is also an intermediate in the production of vanillin from ferulic acid. Vanillic acid inhibits NF-κB activation. Anti-inflammatory, antibacterial, and chemopreventive effects. | ||
| E3625 | Cnidii Fructus Extract | Cnidii Fructus Extract is extracted from Cnidii Fructus, the dry ripe fruit of Cnidium monnieri (L.)Cuss.. Chinese herbal medicine Cnidii Fructus has an outstanding effect on chronic lumbar pain and impotence, also has been used against osteoporosis with high frequency. | ||
| E1351 | NF-κΒ activator 1 | NF-κΒ activator 1(Compound 32) is a potent NF-κΒ activator, with an EC50 of 0.9 μM. | ||
| E3365 | Weigela Grandiflora Fortune Extract | Weigela Grandiflora Fortune Extract is extracted from Weigela Grandiflora Fortune, which decreases the infection-mediated expression of inflammatory mediators by inhibiting the AKT/NF-κB and MAPK signaling pathways. | ||
| S2313 | Indole-3-carbinol | Indole-3-carbinol suppresses NF-κB and IκBα kinase activation, and it is also an inhibitor of WWP1 (E3 ubiquitin ligase with WW domain). |
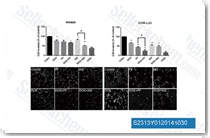
|
|
| S9040 | Maslinic acid | Maslinic Acid (Crategolic Acid, 2α-Hydroxyoleanoic Acid), a Natural Triterpene, exerts a wide range of biological activities, i.e. antitumor, antidiabetic, antioxidant, cardioprotective, neuroprotective, antiparasitic and growth-stimulating. MA significantly suppresses the DNA-binding activity of NF-κB p65 in LPS-induced RAW 264.7 cells. | ||
| S5899 | (R)-(-)-Ibuprofen | (R)-(-)-Ibuprofen ((R)-Ibuprofen) inhibits the activation of NF-κB in response to T-cell stimulation with IC50 of 121.8 μM, and exerts anti-inflammatory and antinociceptive effects. | ||
| S3293 | Gardenoside | Gardenoside is a natural compound extracted from Gardenia fruits, with hepatoprotective properties. Gardenoside inhibits TNF-α, IL-1β, IL-6 and NFκB activation. Gardenoside also has an inhibitory effect on free fatty acids (FFA)-induced cellular steatosis. Gardenoside suppresses the pain in rats model of chronic constriction injury by regulating the P2X3 and P2X7 receptors. | ||
| S9041 | Corosolic acid | Corosolic acid (Glucosol, Colosic acid, 2α-Hydroxyursolic acid) is one of the pentacyclic triterpenoids isolated from Lagerstroemia speciose and has been reported to exhibit anti-cancer and anti-proliferative activities in various cancer cells. | ||
| S3773 | Tyrosol | Tyrosol (4-Hydroxyphenylethanol, 4-Hydroxyphenethyl alcohol, 2-(4-Hydroxyphenyl)ethanol) is an antioxidant that is naturally present in several foods such as wines and green tea and is present most abundantly in olives. Tyrosol is a derivative of phenethyl alcohol. Tyrosol attenuates pro-inflammatory cytokines from cultured astrocytes and NF-κB activation. Anti-oxidative and anti-inflammatory effects. | ||
| E0657 | Rubiadin 1-methyl ether | Rubiadin-1-methyl ether, a natural anthraquinone compound isolated from the root of Morinda officinalis How, can inhibit osteoclastic bone resorption through blocking NF-κB pathway and may be a promising agent for the prevention and treatment of bone diseases characterized by excessive bone resorption. | ||
| E3594 | Radix tetrastigme Extract | Radix Tetrastigme Extract is extracted from Tetrastigma hemsleyanum, which can modulate TLR4/COX-2/NF-κB signaling pathway. | ||
| S9388 | (+)-Praeruptorin A | Praeruptorin A is a coumarin compound naturally occurring in the roots of Peucedanum praeruptorum Dunn., a commonly used traditional Chinese medicine for the treatment of certain respiratory diseases and hypertension. Praeruptorin A exerts anti-inflammatory effects in vitro through inhibition of NF-κB activation. (+)-Praeruptorin A is one of enantiomers. | ||
| S0844 | NDMC101 | NDMC101 inhibits osteoclastogenesis which also ameliorates paw swelling and inflammatory bone destruction, associating with the inhibition of such transcription factors as NF-κB and NFATc1 as well as multiple protein kinases, including p38, ERK, and JNK. | ||
| S9075 | Mulberroside A | Mulberroside A, isolated from the ethanol extract of Morus alba roots, is widely employed as an active ingredient in cosmetic products due to its anti-tyrosinase and anti-oxidant activities. Mulberroside A decreases the expressions of TNF-α, IL-1β and IL-6 and inhibits the activation of NALP3, caspase-1 and NF-κB and the phosphorylation of ERK, JNK and p38. | ||
| S1825 | Erdosteine | Erdosteine (KW-9144,RV 144) is a mucolytic which is used in treatment of excessive viscous mucus. Erdosteine inhibits lipopolysaccharide (LPS)-induced NF-κB activation. | ||
| S3880 | Schisantherin A | Schisantherin A (Gomisin C, Schisanwilsonin H, Arisanschinin K) is a dibenzocyclooctadiene that exhibits anti-tussive, sedative, anti-inflammatory, anti-osteoporotic, neuroprotective, cognition enhancing, and cardioprotective activities. Schisantherin A inhibits p65-NF-κB translocation into the nucleus by IκBα degradation. | ||
| S6679 | C25-140 | C25-140 is a small-molecule inhibitor of TRAF6-Ubc13 interaction. C25-140 directly binds to TRAF6, thereby blocking the interaction of TRAF6 with Ubc13, and as a consequence lowers TRAF6 activity. C25-140 impedes NF-κB activation in various immune and inflammatory signaling pathways also in primary human and murine cells. | ||
| S9417 | Homoplantaginin | Homoplantaginin (Hispidulin-7-glucoside), a main flavonoid from a traditional Chinese medicine Salvia plebeia, has anti-inflammatory and anti-oxidant properties. Homoplantaginin inhibits TNF-α and IL-6 mRNA expression, IKKβ and NF-κB phosphorylation. | ||
| E0694 | Okanin | Okanin, a chalcone found in the flower tea Coreopsis tinctoria, effectively reduces LPS-induced microglial activation by inhibiting the TLR4/NF-κB signalling pathways. It also reduces LPS-induced nitric oxide (NO) production and iNOS expression by increasing HO-1 expression. It exhibits a potential as a nutritional preventive measure for neurodegenerative disorders. | ||
| S9871 | BTYNB | BTYNB (BTYNB IMP1 Inhibitor, MDK6620) is a potent and selective inhibitor of IMP1 binding to c-Myc mRNA. BTYNB downregulates β-TrCP1 mRNA and reduces activation of nuclear transcriptional factors-kappa B (NF-κB). BTYNB disrupts this enhancer function by impairing IGF2 mRNA-binding protein 1 (IGF2BP1)-RNA association. | ||
| S9106 | Eleutheroside E | Eleutheroside E, a principal component of Eleutherococcus senticosus (ES), has anti-inflammatory effects by inhibiting NF-κB and protecting against myocardial infarction. | ||
| S4460 | IMM-H007 | IMM-H007, an adenosine derivative, is an activator of AMP-Activated Protein Kinase (AMPK). IMM-H007 is a potential drug for treating cardiac dysfunction. IMM-H007 negatively regulates endothelium inflammation through inactivating NF-κB and JNK/AP1 signaling. IMM-H007 inhibits ABCA1 (ATP binding cassette subfamily a member 1) degradation and facilitates its cell-surface localization in macrophages, thereby promotes cholesterol efflux. | ||
| S5579 | Chelidonic acid | Chelidonic acid (Jerva acid, Jervaic acid, γ-Pyrone-2,6-dicarboxylic acid) is a secondary metabolite found in several plants with therapeutic potential in allergic disorders in experimental animals. Chelidonic acid inhibits IL-6 production by blocking NF-κB and caspase-1 in HMC-1 cells. Chelidonic acid is also an inhibitor of glutamate decarboxylase with Ki of 1.2 μM. | ||
| S8941 | NIK SMI1 | NIK SMI1 is a highly potent and selective NF-κB-inducing kinase (NIK) inhibitor with Ki of 0.23 nM for NIK-catalyzed hydrolysis of ATP to ADP. | ||
| E1798New | HOIPIN-8 | HOIPIN-8, is a potent inhibitor of linear ubiquitin chain assembly complex (LUBAC) and NF-κB signaling with an IC50 value of 11 nM. It serves as a powerful tool for investigating the physiological and cellular functions of LUBAC. | ||
| E0526 | sappanone A | Sappanone A, a homoisoflavanone isolated from the heartwood of Caesalpinia sappan (Leguminosae), induces HO-1 expression by activating Nrf2 through the p38 MAPK pathway, suppresses LPS-induced NF-κB activation by suppressing the phosphorylation of RelA/p65 at Ser536, exerting its anti-inflammatory effect. | ||
| S3886 | Quinic acid | Quinic acid (Chinic acid, Kinic acid) is a crystalline acid obtained from cinchona bark, coffee beans, and other plant products and made synthetically by hydrolysis of chlorogenic acid. | ||
| S3138 | Methylthiouracil | Methylthiouracil (NSC-193526, NSC-9378,MTU) is an antithyroid agent. Methylthiouracil suppresses the production TNF-α and IL-6, and the activation of NF-κB and ERK1/2. | ||
| S3899 | Hederagenin | Hederagenin (Caulosapogenin, Hederagenol, Hederagenic acid, Astrantiagenin E) is a highly water insoluble triterpenoid compound that can be found in various plants including Hedera helix and Chenopodium quinoa. It exhibits a variety of biological activities, including potent antitumor properties both in vitro and in vivo. Hederagenin inhibits LPS-stimulated expression of iNOS, COX-2, and NF-κB. | ||
| E2661 | Chitosan oligosaccharide | Chitosan oligosaccharide (COS) is an oligomer of β-(1➔4)-linked d-glucosamine, of which actions involve the modulation of several important pathways including the suppression of nuclear factor kappa B (NF-κB) and mitogen-activated protein kinases (MAPK) and the activation of AMP-activated protein kinase (AMPK). | ||
| S1321 | Urolithin B | Urolithin B inhibits NF-κB activity by reducing the phosphorylation and degradation of IκBα. Urolithin B suppresses the phosphorylation of JNK, ERK, and Akt, and enhances the phosphorylation of AMPK. Urolithin B is also a regulator of skeletal muscle mass. | ||
| S3867 | (E)-Cardamonin | (E)-Cardamonin (Alpinetin chalcone, cardamomin) is a naturally occurring chalcone with strong anti-inflammatory activity. It is a novel TRPA1 antagonist with IC50 of 454 nM and also a NF-κB inhibitor. | ||
| S6714 | INH14 | INH14 is an inhibitor of TLR2-mediated NF-κB activation with IC50 values of 8.975 μM and 3.598 μM for IKKα and IKKβ, respectively. | ||
| E1714New | B022 | B022 is a potent and selective NF-κB-inducing kinase (NIK) inhibitor with a Ki of 4.2 nM. B022 protects liver from toxin-induced inflammation, oxidative stress, and injury. | ||
| S3261 | Myrislignan | Myrislignan, a lignan isolated from Myristica fragrans Houtt, possesses anti-inflammatory activities. Myrislignan inhibits interleukin-6 (IL-6) and tumour necrosis factor-α (TNF-α). Myrislignan significantly inhibits the expressions of inducible NO synthase (iNOS) and cyclooxygenase-2 (COX-2) dose-dependently in LPS-stimulated macrophage cells. Myrislignan inhibits the NF-κB signalling pathway activation. | ||
| E3786 | Radix Scrophulariae Extract | Radix Scrophulariae Extract is derived from Radix Scrophulariae, which has anti-apoptotic and anti-inflammatory effects, potentially operating by affecting the mitogen-activated protein kinases (MAPKs) signaling pathway and inhibition of the NF-κB pathway. | ||
| S3872 | Guaiacol | Guaiacol (O-methoxyphenol, 2-hydroxyanisole, O-methylcatechol) is a phenolic natural product first isolated from Guaiac resin and the oxidation of lignin. It is a precursor to various flavorants, such as eugenoland vanillin. Guaiacol, a phenolic compound isolated from Guaiac resin, inhibits LPS-stimulated COX-2 expression and NF-κB activation. Anti-inflammatory activity. | ||
| S4712 | Diethylmaleate | Diethylmaleate (Diethyl ester, Maleic acid diethyl ester) is the diethyl ester of maleic acid and a glutathione-depleting compound that inhibits NF-κB. | ||
| S3298 | Caulophylline (N-Methylcytisine) | Caulophylline (N-Methylcytisine, Caulophyllin, NMC) is a tricyclic quinolizidine alkaloid with anti-inflammatory activities. Caulophylline binds to nicotinic acetylcholine receptors (nAChR) from squid optical ganglia with Kd of 50 nM. Caulophylline significantly reduces myeloperoxidase (MPO) activity, blocks the activation of NF-κB by inhibiting IκB and IKK phosphorylation. | ||
| E3795 | Commelina Communis Extract | Commelina Communis Extract is drawed from Commelina Communis, which has anti-inflammatory effects related to suppression of the NF-κB pathway and can be used to treat chronic inflammation. | ||
| S2371 | Vanillylacetone | Vanillylacetone (NSC 15335) is similar in chemical structure to other flavor chemicals such as vanillin and eugenol and is used as a flavor additive in spice oils and in perfumery to introduce spicy aromas. Vanillylacetone (Zingerone) alleviates oxidative stress and inflammation, down-regulates NF-κB mediated signaling pathways. Zingerone acts as an anti-mitotic agent, and inhibits the growth of neuroblastoma cells. | ||
| S3256 | Tectochrysin | Tectochrysin (Techtochrysin, NSC 80687) is one of the major flavonoids of Alpinia oxyphylla Miquel. Tectochrysin significantly increases the expression of DR3, DR4 and Fas and inhibits activity of NF-κB. Tectochrysin induces apoptotic cell death. | ||
| E0783 | SM-7368 | SM-7368 is a potent NF-κB inhibitor that targets downstream of MAPK p38 activation, also inhibits TNF-α-induced MMP-9 upregulation. | ||
| S9184 | Forsythoside B | Forsythoside B is a phenylethanoid glycoside isolated from the leaves of Lamiophlomis rotata Kudo, a Chinese folk medicinal plant. Forsythoside B has potent neuroprotective effects and has anti-inflammation, antioxidant, and antisepsis properties. Forsythoside B could inhibit TNF-alpha, IL-6, IκB and modulate NF-κB. | ||
| S3609 | Berbamine dihydrochloride | Berbamine (BA, BBM) dihydrochloride, a traditional Chinese medicines extracted from Berberis amurensis (xiaoboan), is a novel inhibitor of bcr/abl fusion gene with potent anti-leukemia activity and also an inhibitor of NF-κB. Berbamine (BA, BBM) dihydrochloride induces apoptosis in human myeloma cells and inhibits the growth of cancer cells by targeting Ca²⁺/calmodulin-dependent protein kinase II (CaMKII). | ||
| S3299 | Demethyleneberberine | Demethyleneberberine (DMB), a component of Cortex Phellodendri Chinensis (CPC), significantly alleviates the weight loss and diminishes myeloperoxidase (MPO) activity, while significantly reduces the production of pro-inflammatory cytokines, such as interleukin (IL)-6 and tumor necrosis factor-α (TNF-α), and inhibits the activation of NF-κB signaling pathway. Demethyleneberberine (DMB) potentially ameliorates NAFLD (Non-alcoholic fatty liver disease) by activating AMPK pathways. | ||
| E0211 | Dihydroberberine | Dihydroberberine, a hydrogenated derivative of Berberine (BBR), exerts anti-inflammatory effect via dual modulation of NF-κB and MAPK signaling pathways. |
||
| S1013 | Bortezomib | Bortezomib is a potent 20S proteasome inhibitor with Ki of 0.6 nM. It exhibits favorable selectivity towards tumor cells over normal cells. Bortezomib (PS-341) inhibits NF-κB and induces ERK phosphorylation to suppress cathepsin B and inhibit the catalytic process of autophagy in ovarian cancer and other solid tumors. |

|
|
| S1623 | Acetylcysteine (N-acetylcysteine) | Acetylcysteine (N-acetyl-l-cysteine, NAC,N-acetylcysteine) is a ROS(reactive oxygen species) inhibitor that antagonizes the activity of proteasome inhibitors. It is also a tumor necrosis factor production inhibitor. Acetylcysteine(N-acetyl-l-cysteine) suppresses TNF-induced NF-κB activation through inhibition of IκB kinases. Acetylcysteine(N-acetyl-l-cysteine) induces apoptosis via the mitochondria-dependent pathway. Acetylcysteine(N-acetyl-l-cysteine) inhibits ferroptosis and virus replication.Solutions are unstable and should be fresh-prepared. |

|
|
| S7351 | JSH-23 | JSH-23 is an inhibitor of NF-κB transcriptional activity with IC50 of 7.1 μM in RAW 264.7 cell line. |

|
|
| S4902 | QNZ (EVP4593) | QNZ (EVP4593) shows potent inhibitory activity toward both NF-κB activation and TNF-α production with IC50 of 11 nM and 7 nM in Jurkat T cells, respectively. |

|
|
| S3633 | PDTC (Pyrrolidinedithiocarbamate ammonium) | PDTC (Pyrrolidinedithiocarbamate ammonium) is a potent nuclear factor-κB (NF-κB) inhibitor that inhibits IκB phosphorylation, blocks NF-κB translocation to the nucleus and reduces the expression of downstream cytokines. | ||
| S2824 | TPCA-1 | TPCA-1 (GW683965) is an inhibitor of IKK-2 with IC50 of 17.9 nM in a cell-free assay, inhibits NF-κB pathway, exhibits 22-fold selectivity over IKK-1. TPCA-1 is also an inhibitor of STAT3 and enhances apoptosis. |

|
|
| S7273 | SC75741 | SC75741 is a potent NF-κB inhibitor with EC50 of 200 nM. SC75741 efficiently blocks influenza virus propagation. |

|
|
| S7414 | Caffeic Acid Phenethyl Ester | Caffeic acid phenethyl ester (CAPE, Phenylethyl Caffeate) is a potent and specific inhibitor of NF-κB activation, and also displays antioxidant, immunomodulatory and antiinflammatory activities. |

|
|
| S1848 | Curcumin | Curcumin (Diferuloylmethane, Natural Yellow 3, Turmeric yellow) is the principal curcuminoid of the popular Indian spice turmeric, which is a member of the ginger family (Zingiberaceae). It is an inhibitor of p300 histone acetylatransferase(IC50~25 μM)and Histone deacetylase (HDAC); activates Nrf2 pathway and supresses the activation of NF-κB. Curcumin induces mitophagy, autophagy, apoptosis, and cell cycle arrest with antitumor activity. Curcumin reduces renal damage associated with rhabdomyolysis by decreasing ferroptosis-mediated cell death. Curcumin exhibits anti-infective properties against various human pathogens like the influenza virus, hepatitis C virus, HIV and so on. |

|
|
| S8341 | TAK-243 (MLN7243) | TAK-243 (MLN7243) is a potent, mechanism-based small-molecule inhibitor of the ubiquitin activating enzyme (UAE) with an IC50 of 1 ± 0.2 nM in the UBCH10 E2 thioester assay. It has minimal inhibitory activity in a panel of kinase and receptor assays, as well as on human carbonic anhydrase type I and type II. TAK-243 (MLN7243) induces ER stress, abrogates NFκB pathway activation and promotes apoptosis. | ||
| S3604 | Triptolide | Triptolide is a diterpene triepoxide, immunosuppresive agent extracted from the Chinese herb Tripterygium wilfordii. It functions as a NF-κB inhibitor with dual actions by disruption of p65/CBP interaction and by reduction of p65 protein. Triptolide (PG490) abrogates the transactivation function of heat shock transcription factor 1 (HSF1). Triptolide inhibits MDM2 and induces apoptosis through a p53-independent pathway. |

|
|
| S8279 | Shikonin | Shikonin, a potent and specific Pyruvate kinase M2 (PKM2) inhibitor, is a major component of zicao (purple gromwell, the dried root of Lithospermum erythrorhizon), a Chinese herbal medicine with various biological activities. It is also an inhibitor of TMEM16A chloride channel activity using cell-based fluorescent-quenching assay. Shikonin exerts an anti-inflammatory effect by inhibiting tumor necrosis factor-α (TNF-α) and prevents activation of nuclear factor-κB (NF-κB) pathway via proteasome inhibition. | ||
| S8078 | Bardoxolone Methyl | Bardoxolone Methyl (RTA 402, TP-155, NSC 713200, CDDO Methyl Ester, CDDO-Me) is an IKK inhibitor, showing potent proapoptotic and anti-inflammatory activities; Also a potent Nrf2 activator and nuclear factor-κB (NF-κB) inhibitor. Bardoxolone Methyl abrogates ferroptosis. Bardoxolone methyl induces apoptosis and autophagy in cancer cells. |

|
|
| S2290 | DHA (Dihydroartemisinin) | DHA (Dihydroartemisinin) is a semi-synthetic derivative of artemisinin and isolated from the traditional Chinese herb Artemisia annua. Dihydroartemisinin induces autophagy and apoptosis by suppressing NF-κB activation. |

|
|
| S2341 | (-)-Parthenolide | (-)-Parthenolide, an inhibitor of the Nuclear Factor-κB Pathway, specifically depletes HDAC1 protein without affecting other class I/II HDACs; Also promotes the ubiquitination of MDM2 and activates p53 cellular functions. |
-Parthenolide-S234101W0220161110.gif)
|
|
| S1576 | Sulfasalazine | Sulfasalazine is a sulfa derivative of mesalazine, used as an anti-inflammatory agent to treat bowel disease and rheumatoid arthritis. Sulfasalazine is a potent and specific inhibitor of nuclear factor kappa B (NF-κB), TGF-β and COX-2. Sulfasalazine induces ferroptosis, apoptosis and autophagy. | ||
| S5771 | Sulforaphane | Sulforaphane is a naturally occurring isothiocyanate derived from the consumption of cruciferous vegetables, such as broccoli, cabbage, and kale. It is an inducer of Nrf2. Sulforaphane is also an inhibitor of histone deacetylase (HDAC) and NF-κB. Sulforaphane increases heme oxygenase-1 (HO-1) and reduces the levels of reactive oxygen species (ROS). Sulforaphane induces cell cycle arrest and apoptosis., | ||
| S2261 | Andrographolide | Andrographolide is a labdane diterpenoid that is the main bioactive component of the medicinal plant Andrographis paniculata. | ||
| S2382 | Evodiamine | Evodiamine (Isoevodiamine), an alkaloid extract from the fruit of Evodiae Fructus exhibits antitumor activities against the human tumor cells. It is shown to inhibit NF-κB activation through suppression of IkB kinase activity. |

|
|
| S8587 | Withaferin A (WFA) | Withaferin A (WFA) potently inhibits NF-κB activation by preventing the tumor necrosis factor-induced activation of IκB kinase β via a thioalkylation-sensitive redox mechanism. Withaferin A binds to the intermediate filament (IF) protein, vimentin with antitumor and antiangiogenesis activity. Withaferin A is a steroidal lactone isolated from Withania somnifera. | ||
| S8483 | CBL0137 HCl | CBL0137 (CBLC137, Curaxin 137) HCl activates p53 and inhibits NF-κB with EC50s of 0.37 μM and 0.47 μM in the cell-based p53 and NF-kB reporter assays, respectively. It also inhibits histone chaperone FACT (facilitates chromatin transcription complex). | ||
| S0507 | CBL0137 | CBL0137 (Curaxin-137) is an inhibitor of the histone chaperone FACT (facilitates chromatin transcription) that simultaneously suppresses NF-κB and activates p53 with EC50 of 0.47 μM and 0.37 μM, respectively. | ||
| S2321 | Magnolol | Magnolol (NSC 293099) is a bioactive lignin found in the bark of the Houpu magnolia (Magnolia officinalis) which shows antifungal properties. It can block TNF-α-induced NF-κB activation. |

|
|
| S7672 | Omaveloxolone (RTA-408) | Omaveloxolone (RTA-408) is a synthetic triterpenoid that activates the cytoprotective transcription factor Nrf2 and inhibits NF-κB signaling. Phase 2. | ||
| S9141 | Berbamine | Berbamine (BA), a traditional Chinese medicines extracted from Berberis amurensis (xiaoboan), is a novel inhibitor of bcr/abl fusion gene with potent anti-leukemia activity and also an inhibitor of NF-κB. Berbamine (BA) induces apoptosis in human myeloma cells and inhibits the growth of cancer cells by targeting Ca²⁺/calmodulin-dependent protein kinase II (CaMKII). | ||
| S5453 | Hyperoside | Hyperoside (Hyperin, Quercetin 3-galactoside), a naturally occuring flavonoid compound, exerts multiple bioactivities, including myocardial protection, anti-redox, and anti-inflammatory activities.Hyperoside can inhibit activation of the NF-κB signaling pathway. | ||
| S5144 | Neferine | Neferine ((R)-1,2-Dimethoxyaporphine), a natural component of Nelumbo nucifera, has antitumor efficiency. Neferine induces apoptosis in renal cancer cells. Neferine prevents autophagy through activation of Akt/mTOR pathway and Nrf2 in muscle cells. Neferine strongly inhibits NF-κB activation. Neferine possesses a number of therapeutic effects such as anti-diabetic, anti-aging, anti-microbial, anti-thrombotic, anti-arrhythmic, anti-inflammatory and even anti-HIV. | ||
| S3901 | Astragaloside IV | Astragaloside IV (AST-IV, AS-IV) is a bioactive saponin first isolated from the dried plant roots of the genus Astragalus, which is used in traditional Chinese medicine. It has various effect on the cardiovascular, immune, digestive, and nervous systems. AS-IV suppresses activation of p-Akt, p-mTOR, p-NF-κB and p-Erk1/2. | ||
| S6671 | SN50 | SN50 (NF-κB SN50), a cell-permeable NF-κB inhibitory peptide, is composed of the signal peptide of Kaposi fibroblast growth factor.SN50 inhibits the activation of NF-κB and attenuates ventilator-induced lung injury. | ||
| S7445 | APX-3330 | APX-3330 (E3330) is a potent and selective APE1(Ref-1) inhibitor, which suppressed NF-kappa B DNA-binding activity. | ||
| S3931 | Ginsenoside Rd | Ginsenoside Rd (Panaxoside Rd, Sanchinoside Rd), a minor ginseng saponin, has several pharmacological activities such as immunosuppressive activity, anti-inflammatory activity, immunological adjuvant, anti-cancer activity and wound-healing activity. Ginsenoside Rd inhibits TNFα-induced NF-κB transcriptional activity with an IC50 of 12.05±0.82 μM in HepG2 cells. Ginsenoside Rd inhibits expression of COX-2 and iNOS mRNA. Ginsenoside Rd also inhibits Ca2+ influx. Ginsenoside Rd inhibits CYP2D6, CYP1A2, CYP3A4, and CYP2C9, with IC50s of 58.0±4.5 μM, 78.4±5.3 μM, 81.7±2.6 μM, and 85.1±9.1 μM, respectively. | ||
| S3771 | Stachydrine | Stachydrine (Proline betaine, L-stachydrine, Methyl hygrate betaine) is a quaternary ammonium derivative of proline that occurs widely in Medicago species. It is an osmoprotective compound found in urine. Stachydrine is a major constituent of Chinese herb leonurus heterophyllus sweet used to promote blood circulation and dispel blood stasis. Stachydrine can inhibit the NF-κB signal pathway. | ||
| S3874 | Curcumenol | Curcumenol, a sesquiterpene isolated from Curcuma zedoaria, is known to possess a variety of health and medicinal values which includes neuroprotection, anti-inflammatory, anti-tumor and hepatoprotective activities. It inhibits NF-κB activation by suppressing the nuclear translocation of the NF-κB p65 subunit and blocking IκBα phosphorylation and degradation. | ||
| S3924 | Ginsenoside Rb1 | Ginsenoside Rb1 (Gypenoside Ⅲ) is a protopanaxadiol that has diverse in vitro and in vivo effects, including neuroprotective, anti-inflammatory, and anti-obesity actions. Ginsenoside Rb1, a main constituent of the root of Panax ginseng, inhibits Na+, K+-ATPase activity with IC50 of 6.3±1.0 μM. Ginsenoside also inhibits IRAK-1 activation and phosphorylation of NF-κB p65. Ginsenoside Rb1 reduces the expressions of TLR3, TLR4 and TRAF-6, and down-regulates the levels of TNF-α, IFN-β and iNOS. | ||
| S4073 | Sodium 4-Aminosalicylate | Sodium 4-Aminosalicylate is an antibiotic used to treat tuberculosis via NF-κB inhibition and free radical scavenging. |

|
|
| E0796 | NF-κB-IN-1 | NF-κB-IN-1 is a potent NF-κB signaling pathway inhibitor, which acts by directly inhibiting inhibitory κB kinase (IKK). | ||
| S9193 | Aristolochic acid A | Aristolochic acid A (Aristolochic Acid I, Aristolochin, Aristolochine, TR 1736) is a carcinogenic, mutagenic, and nephrotoxic agent extracted from the flowering plant family Aristolochiaceae.Aristolochic acid A significantly reduces both activator protein 1 (AP-1) and nuclear factor-κB (NF-κB) activities. | ||
| S7428 | Rocaglamide | Rocaglamide (Roc-A), isolated from Aglaia species, is a potent inhibitor of heat shock factor 1 (HSF1) activation with IC50 of ~50 nM for HSF1. Rocaglamide inhibits the function of the translation initiation factor eIF4A. Rocaglamide also inhibits NF-κB activity. Rocaglamide exhibits anti-tumor activity. | ||
| S2416 | Chondroitin sulfate | Chondroitin sulfate (CS) is a major component of the extracellular matrix (ECM) of many connective tissues, including cartilage, bone, skin, ligaments and tendons. It reduces the IL-1β-induced nuclear factor-kB (NF-κB) translocation in chondrocytes in vitro. | ||
| S9170 | Engeletin | Engeletin (Dihydrokaempferol 3-rhamnoside), a bioactive flavonoid, has multiple biological activities such as anti-diabetic and anti-inflammatory effects. engeletin againsts LPS-stimulated endometritis in mice via negative regulation of pro-inflammatory mediators via the TLR4-regulated NF-κB pathway. | ||
| S3923 | Ginsenoside Rg1 | Ginsenoside Rg1 (Ginsenoside A2, Panaxoside A, Panaxoside Rg1, Sanchinoside C1, Sanchinoside Rg1), one of the major active components of ginseng, is identified as a protopanaxatriol-type and has pharmacological actions such as neuroprotective and anti-tumor effects on various cancer types. Ginsenoside Rg1 reduces cerebral Aβ levels and NF-κB nuclear translocation. | ||
| S3137 | Sodium salicylate | Sodium salicylate(2-Hydroxybenzoic acid Sodium salt,Salicylic acid Sodium salt) is used in medicine as an analgesic and antipyretic. | ||
| S4798 | Benfotiamine | Benfotiamine (S-Benzoylthiamine O-monophosphate) is a synthetic S-acyl derivative of thiamine (vitamin B1) and has been investigated for the treatment and prevention of Diabetic Nephropathy and Diabetes Mellitus, Type 2. Benfotiamine suppresses oxidative stress-induced NF-κB activation and prevents the bacterial endotoxin-induced inflammation. | ||
| S5640 | Ethyl caffeate | Ethyl caffeate, a naturally occurring compound found in Bidens pilosa, suppresses NF-kappaB activation and its downstream inflammatory mediators, iNOS, COX-2 and PGE2 in vitro. | ||
| S9295 | Dauricine | Dauricine, a plant metabolite isolated from the Asian vine Menispermum dauricum, plays a variety of biological roles in the human body, from inhibiting cancer cell growth to blocking cardiac transmembrane Na+, K+, and Ca2+ ion currents.Dauricine induces apoptosis, inhibits proliferation and invasion through inhibiting NF‐κB signaling pathway in colon cancer cells. | ||
| S3770 | Sodium Aescinate | Sodium Aescinate (SA, Escin Sodium Salt) is a widely-applied triterpene saponin product derived from horse chestnut seeds, possessing vasoactive and organ-protective activities with oral or injection administration in the clinic. Sodium aescinate is a triterpene saponin derived from Aesculus hippocastanum seeds, with anti-inflammatory and antioxidant activities. Sodium aescinate inhibits hepatocellular carcinoma growth by targeting CARMA3/NF-κB pathway. | ||
| S9374 | 2',5'-Dihydroxyacetophenone | 2',5'-Dihydroxyacetophenone (DHAP, 2-Acetylhydroquinone, Quinacetophenone) is a mixture of dihydroxyacetophenone isomers is used in food flavouring. 2',5'-Dihydroxyacetophenone significantly inhibits NO production via the suppression of iNOS expression. 2',5'-Dihydroxyacetophenone significantly decreases levels of the pro-inflammatory cytokines TNF-α and IL-6 by blocking the ERK1/2 and NF-κB signaling pathways. | ||
| S3871 | Muscone | Muscone (3-Methylcyclopentadecanone, Methylexaltone), a flavouring ingredient, is an organic compound that is the primary contributor to the odor of musk and also a potent anti-inflammatory agent. Muscone significantly downregulats the levels of LPS-induced inflammatory cytokines and inhibits NF-κB and NLRP3 inflammasome activation in BMDMs. Muscone remarkably decreases the levels of inflammatory cytokines (IL-1β, TNF-α and IL-6). | ||
| S7637 | DTP3 | DTP3 is a selective GADD45β/MKK7 (growth arrest and DNA-damage-inducible β/mitogen-activated protein kinase kinase 7) inhibitor and is able to restore MKK7/JNK activation. DTP3 inhibits cancer-selective NF-κB survival pathway. | ||
| S4763 | 4-Hydroxychalcone | 4-Hydroxychalcone (P-Cinnamoylphenol) is a chalcone metabolite with diverse biological activities. It inhibits TNFα-induced NF-κB pathway activation in a dose-dependent manner and also activates BMP signaling. | ||
| S3808 | Mangiferin | Mangiferin (Alpizarin, Chinomin, Hedysarid) is a bioactive compound that demonstrates many health perspectives and has been used to prepare medicinal and food supplements. Mangiferin is a Nrf2 activator. Mangiferin suppresses nuclear translocation of the NF-κB subunits p65 and p50. | ||
| S3810 | Scutellarin | Scutellarin (Breviscapine, Breviscapin, Scutellarein-7-glucuronide), the major active principal flavonoids extracted from the Chinese herbal medicines Scutellaria baicalensis and Erigeron breviscapus (Vant.) Hand-Mazz, has many pharmacological effects, such as antioxidant, antitumor, antiviral, and antiinflammatory activities. Scutellarin can down-regulates the STAT3/Girdin/Akt signaling in HCC cells, and inhibits RANKL-mediated MAPK and NF-κB signaling pathway in osteoclasts. | ||
| S3607 | Sarsasapogenin | Sarsasapogenin (SAR, Parigenin) is a steroidal sapogenin. It can provoke the generation of reactive oxygen species and activate unfolded protein response (UPR) signaling pathways. SAR potently inhibits NF-κB and MAPK activation, as well as IRAK1, TAK1, and IκBα phosphorylation in LPS-stimulated macrophages. | ||
| S3811 | Ginsenoside Re | Ginsenoside Re (Ginsenoside B2, Panaxoside Re, Sanchinoside Re, Chikusetsusaponin Ivc), an extract from Panax notoginseng, is a major ginsenoside in ginseng and belongs to 20(S)-protopanaxatriol group. It has diverse in vitro and in vivo effects, including vasorelaxant, antioxidant, antihyperlipidemic, and angiogenic actions. Ginsenoside Re decreases the β-amyloid protein (Aβ). Ginsenoside Re plays a role in antiinflammation through inhibition of JNK and NF-κB. | ||
| S9502 | Madecassic acid | Madecassic acid (Brahmic acid), a natural triterpene first isolated from C. asiatica, has diverse anti-inflammatory and anti-diabetic effects, blocking NF-κB activation in macrophages and causing by iNOS, COX-2, TNF-alpha, IL-1beta and IL-6 inhibition. | ||
| S0454 | Adjudin | Adjudin (AF-2364), a potent male contraceptive, is a potent blocker of Cl⁻ channels. Adjudin exhibits anti-inflammatory properties by suppression of NF-κB p65 nuclear translocation and DNA binding activity as well as ERK MAPK phosphorylation. | ||
| S3150 | Articaine HCl | Articaine (Ultracaine) is a dental local anesthetic which contains an additional ester group that is metabolized by estearases in blood and tissue. | ||
| S3939 | 4'-Methoxyresveratrol | 4-Methoxyresveratrol is a stibenoid found in the Chinese herb Gnetum cleistostachyum. 4'-Methoxyresveratrol alleviates AGE-induced inflammation through suppressing RAGE-mediated MAPK/NF-κB signaling pathway and NLRP3 inflammasome activation. | ||
| E0212 | Cornuside | Cornuside is a bisiridoid glucoside compound isolated from the fruit of Cornus officinalis SIEB. et ZUCC. Cornuside suppresses lipopolysaccharide-induced inflammatory mediators by inhibiting NF-κB activation in RAW 264.7 macrophages. Cornuside attenuates apoptosis in rat cortical neurons. |
||
| E3034 | Cynanchi Atrati Extract | Cynanchi Atrati Extract is extracted from Cynanchi atrati Radix, has been traditionally used as an anti-inflammatory agent, and has a potential to inhibit IKK-mediated NF-κB activation, which is valuable for modulating inflammatory or cancerous conditions. | ||
| S0002 | IAXO-102 | IAXO-102 is an antagonist of TLR4 targeting both MD-2 and CD14 co-receptors. IAXO-102 inhibits MAPK and p65 NF-κB phosphorylation and downregulates the expression of TLR4 dependent proinflammatory proteins. IAXO-102 inhibits experimental abdominal aortic aneurysm (AAA) development. | ||
| S6899 | Licochalcone D | Licochalcone D (Lico D, LCD, LD), a flavonoid isolated from a Chinese medicinal plant Glycyrrhiza inflata, has antioxidant, anti-inflammatory and anti-cancer properties. Licochalcone D inhibit phosphorylation of NF-κB p65 in LPS signaling pathway. Licochalcone D inhibits JAK2, EGFR and Met (c-Met) activities and induces ROS-dependent apoptosis. Licochalcone D also induces caspases activation and poly (ADP-ribose) polymerase (PARP) cleavage. | ||
| S0911 | Hypaphorine | Hypaphorine which is abundantly found in vaccaria semen, counteracts inflammation via inhibition of ERK or/and NFκB signaling pathways. | ||
| S3223 | L-Quebrachitol | L-Quebrachitol (L-QCT), a natural product isolated from many plants, promotes proliferation and cell DNA synthesis. L-Quebrachitol upregulates bone morphogenetic protein-2 (BMP-2) and runt-related transcription factor-2 (Runx2) and regulatory genes associated with mitogen-activated protein kinase (MAPK) and Wnt/β-catenin signaling pathway, while down-regulating the receptor activator of the nuclear factor-κB(NF-κB) ligand (RANKL) mRNA level. | ||
| S3998 | (+)-α-Lipoic acid | (+)-α-Lipoic acid ((R)-(+)-α-Lipoic acid, α-Lipoic acid, Alpha-Lipoic acid), a physiological form of thioctic acid, is a strong antioxidant that relieves diabetic neuropathic symptoms. It shows superior antioxidative effects to its racemate. R(+)-α-lipoic acid is an essential cofactor of mitochondrial enzyme complexes. R(+)-α-lipoic acid inhibits NF-κB-dependent HIV-1 LTR activation. | ||
| S0915 | Morusin | Morusin (Mulberrochromene) is an inhibitor of NF-κB and STAT3. Morusin is a prenylated flavonoid isolated from Marsdenia australis with antitumor, antioxidant, and anti-bacteria properties. | ||
| S9208 | Ginsenoside Rb3 | Ginsenoside Rb3, extracted from the plant Panax ginseng, plays important roles in cardiovascular diseases, including myocardial ischemia-reperfusion (I/R) injury. Ginsenoside Rb3 (0.1-10 μM) is tested for inhibition of tumor necrosis factor-α (TNF)-induced nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) luciferase reporter activity using a human kidney 293T cell-based assay. Ginsenoside Rb3 shows the significant activity with an IC50 of 8.2 μM. Ginsenoside Rb3 also inhibits the induction of cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) messenger Ribonucleic acid (mRNA) in a dose-dependent manner after HepG2 cells have been treated with TNF-α (10 ng/mL). | ||
| S0923 | Isoliquiritin apioside | Isoliquiritin apioside (ISLA, ILA), a component isolated from Glycyrrhizae radix rhizome (GR), significantly decreases PMA-induced increases in matrix metalloproteinase (MMP) activities and suppresses PMA-induced activation of mitogen-activated protein kinase (MAPK) and NF-κB. Isoliquiritin apioside possesses anti-metastatic and anti-angiogenic abilities in malignant cancer cells and ECs, with no cytotoxicity. | ||
| S8961 | Alobresib (GS-5829) | Alobresib (GS-5829) is a novel BET inhibitor that represents a highly effective therapeutics agent against recurrent/chemotherapy-resistant USC-overexpressing c-Myc. Alobresib (GS-5829) inhibits CLL cell proliferation and induces leukemia cell apoptosis through deregulation of key signaling pathways, such as BLK, AKT, ERK1/2, and MYC. Alobresib (GS-5829) also inhibits NF-κB signaling. | ||
| E4443New | S-Nitrosoglutathione | S-nitrosoglutathione (GSNO, RVC-588, S-Nitroso-L-glutathione,Nitrosoglutathione) is an amino acid and nitric oxide donor that, under physiological conditions, spontaneously releases nitric oxide. S-nitrosoglutathione (GSNO) selectively inhibits platelet and NF-κB activation. It induces apoptosis in T cells in low concentrations (< 0.6 mM) while protecting T cells from apoptosis in higher concentrations (1-2 mM). | ||
| S0931 | Jaceosidin | Jaceosidin, a flavonoid isolated from Artemisia vestita, possesses anti-tumor and anti-proliferative activities in many cancer cells. Jaceosidin induces apoptosis, activates Bax and down-regulates Mcl-1 and c-FLIP expression. Jaceosidin inhibits COX-2 expression and NF-κB activation. | ||
| S3243 | Zeaxanthin | Zeaxanthin, the carotenoid alcohol participates in the xanthophyll cycle, activates the extrinsic apoptosis pathway which induces apoptosis on uveal melanoma cells with IC50 value 40.8 µM. | ||
| S3231 | UCB-9260 | UCB-9260 is an orally active inhibitor that inhibits TNF signalling by stabilising an asymmetric form of the trimer. UCB-9260 binds TNF with Kd of 13 nM. UCB-9260 also inhibits NF-κB with IC50 of 202 nM after TNF stimulation. | ||
| S8989 | Xanthatin | Xanthatin is a sesquiterpene lactone isolated from Xanthium strumarium leaves, which can inhibit the nuclear factor kappa-B (NF-κB), mitogen-activated protein kinase (MAPK) and signal transducer and activator of transcription (STATs) signaling pathways. | ||
| S3816 | Dehydroevodiamine | Dehydroevodiamine (DHED), a constituent of Evodia rutaecarpa, has various biological effects such as hypotensive, negative chronotropic, ion channel depressant, inhibition of nitric oxide production and cerebral blood flow enhancing activities. Dehydroevodiamine inhibits LPS-induced iNOS, COX-2, prostaglandin E2 (PGE2) and nuclear factor-kappa B (NF-κB) expression in murine macrophage cells. | ||
| S9244 | 8-O-acetyl shanzhiside methyl ester | 8-O-acetyl shanzhiside methyl ester (Barlerin, ND01), isolated from the leaves of Lamiophlomis rotata Kudo, promotes angiogenesis, which leads to the improvement of functional outcome after stroke.8-O-Acetyl shanzhiside methyl ester can inhibts NF-κB. | ||
| S3384 | (E/Z)-IT-603 | (E/Z)-IT-603 is a mixture of E-IT-603 and Z-IT-603. IT-603 is a NF-κB family member c-Rel inhibitor with an IC50 of 3 μM in electrophoretic mobility shift assay (EMSA). | ||
| P1238New | SN 52 | SN52, a cell-permeable peptide containing the p50 NLS, is a selective inhibitor of NF-κB pathway. It selectively and completely blocks the ionizing radiation (IR)-induced nuclear import of p52 and RelB. It also enhances the radiosensitivity of prostate cancer cells. | ||
| E3652 | Rehmanniae radix praeparata Extract | Rehmanniae radix praeparata Extract is extracted from Rehmanniae radix praeparata, which can improve diabetes through AMPK-mediated NF-κB/NLRP3 signaling pathway. | ||
| E3654 | Althaea rosea Extract(root) | Althaea rosea Extract(root) is extracted from the root of Althaea rosea Extract, which can inhibit NF-κB. | ||
| E1575New | EN450 | EN450 is a cysteine-reactive covalent molecular glue degrader targeting NF-κB. It interacts with allosteric C111 in the E2 ubiquitin ligase UBE2D that induces the ternary complex formation between UBE2D and NFKB1. It exhibits anti-proliferative effects through a Cullin E3 ligase and proteasome-dependent mechanism. | ||
| S0781 | IQ 3 | IQ 3 is a specific c-Jun N-terminal kinase (JNK) inhibitor with Kd of 66 nM, 240 nM and 290 nM for JNK3, JNK1 and JNK2, respectively. IQ 3 inhibits LPS-induced NF-κB/AP1 transcriptional activity in THP1-Blue cells with IC50 of 1.4 μM. IQ 3 also inhibits TNF-α and IL-6 production in vitro. | ||
| E0022 | Ophiopogonin D | Ophiopogonin D (OJV-VI, Deacetylophiopogonin C) is a steroidal glycoside isolated from Chinese herb Radix ophiopogonis and shows anti-tumor property. Ophiopogonin D could suppress TGF-β1-mediated metastatic behavior of MDA-MB-231 cells by regulating ITGB1/FAK/Src/AKT/β-catenin/MMP-9 signaling axis. Ophiopogonin D attenuates PM2.5-induced inflammation via suppressing the AMPK/NF-κB pathway in mouse pulmonary epithelial cells. | ||
| S4937 | 4'-Hydroxychalcone | 4'-Hydroxychalcone (P-Cinnamoylphenol), found in herbs and spices and tea, is a member of the class of compounds known as retrochalcones. It has diverse biological activities, inhibiting TNFα-induced NF-κB pathway activation in a dose-dependent manner and activating BMP signaling. | ||
| S5343 | Vanillic acid | Vanillic acid (4-hydroxy-3-methoxybenzoic acid) is a flavoring agent which is also an intermediate in the production of vanillin from ferulic acid. Vanillic acid inhibits NF-κB activation. Anti-inflammatory, antibacterial, and chemopreventive effects. | ||
| E3365 | Weigela Grandiflora Fortune Extract | Weigela Grandiflora Fortune Extract is extracted from Weigela Grandiflora Fortune, which decreases the infection-mediated expression of inflammatory mediators by inhibiting the AKT/NF-κB and MAPK signaling pathways. | ||
| S2313 | Indole-3-carbinol | Indole-3-carbinol suppresses NF-κB and IκBα kinase activation, and it is also an inhibitor of WWP1 (E3 ubiquitin ligase with WW domain). |

|
|
| S9040 | Maslinic acid | Maslinic Acid (Crategolic Acid, 2α-Hydroxyoleanoic Acid), a Natural Triterpene, exerts a wide range of biological activities, i.e. antitumor, antidiabetic, antioxidant, cardioprotective, neuroprotective, antiparasitic and growth-stimulating. MA significantly suppresses the DNA-binding activity of NF-κB p65 in LPS-induced RAW 264.7 cells. | ||
| S5899 | (R)-(-)-Ibuprofen | (R)-(-)-Ibuprofen ((R)-Ibuprofen) inhibits the activation of NF-κB in response to T-cell stimulation with IC50 of 121.8 μM, and exerts anti-inflammatory and antinociceptive effects. | ||
| S3293 | Gardenoside | Gardenoside is a natural compound extracted from Gardenia fruits, with hepatoprotective properties. Gardenoside inhibits TNF-α, IL-1β, IL-6 and NFκB activation. Gardenoside also has an inhibitory effect on free fatty acids (FFA)-induced cellular steatosis. Gardenoside suppresses the pain in rats model of chronic constriction injury by regulating the P2X3 and P2X7 receptors. | ||
| S3773 | Tyrosol | Tyrosol (4-Hydroxyphenylethanol, 4-Hydroxyphenethyl alcohol, 2-(4-Hydroxyphenyl)ethanol) is an antioxidant that is naturally present in several foods such as wines and green tea and is present most abundantly in olives. Tyrosol is a derivative of phenethyl alcohol. Tyrosol attenuates pro-inflammatory cytokines from cultured astrocytes and NF-κB activation. Anti-oxidative and anti-inflammatory effects. | ||
| E0657 | Rubiadin 1-methyl ether | Rubiadin-1-methyl ether, a natural anthraquinone compound isolated from the root of Morinda officinalis How, can inhibit osteoclastic bone resorption through blocking NF-κB pathway and may be a promising agent for the prevention and treatment of bone diseases characterized by excessive bone resorption. | ||
| S9388 | (+)-Praeruptorin A | Praeruptorin A is a coumarin compound naturally occurring in the roots of Peucedanum praeruptorum Dunn., a commonly used traditional Chinese medicine for the treatment of certain respiratory diseases and hypertension. Praeruptorin A exerts anti-inflammatory effects in vitro through inhibition of NF-κB activation. (+)-Praeruptorin A is one of enantiomers. | ||
| S9075 | Mulberroside A | Mulberroside A, isolated from the ethanol extract of Morus alba roots, is widely employed as an active ingredient in cosmetic products due to its anti-tyrosinase and anti-oxidant activities. Mulberroside A decreases the expressions of TNF-α, IL-1β and IL-6 and inhibits the activation of NALP3, caspase-1 and NF-κB and the phosphorylation of ERK, JNK and p38. | ||
| S1825 | Erdosteine | Erdosteine (KW-9144,RV 144) is a mucolytic which is used in treatment of excessive viscous mucus. Erdosteine inhibits lipopolysaccharide (LPS)-induced NF-κB activation. | ||
| S3880 | Schisantherin A | Schisantherin A (Gomisin C, Schisanwilsonin H, Arisanschinin K) is a dibenzocyclooctadiene that exhibits anti-tussive, sedative, anti-inflammatory, anti-osteoporotic, neuroprotective, cognition enhancing, and cardioprotective activities. Schisantherin A inhibits p65-NF-κB translocation into the nucleus by IκBα degradation. | ||
| S6679 | C25-140 | C25-140 is a small-molecule inhibitor of TRAF6-Ubc13 interaction. C25-140 directly binds to TRAF6, thereby blocking the interaction of TRAF6 with Ubc13, and as a consequence lowers TRAF6 activity. C25-140 impedes NF-κB activation in various immune and inflammatory signaling pathways also in primary human and murine cells. | ||
| S9417 | Homoplantaginin | Homoplantaginin (Hispidulin-7-glucoside), a main flavonoid from a traditional Chinese medicine Salvia plebeia, has anti-inflammatory and anti-oxidant properties. Homoplantaginin inhibits TNF-α and IL-6 mRNA expression, IKKβ and NF-κB phosphorylation. | ||
| E0694 | Okanin | Okanin, a chalcone found in the flower tea Coreopsis tinctoria, effectively reduces LPS-induced microglial activation by inhibiting the TLR4/NF-κB signalling pathways. It also reduces LPS-induced nitric oxide (NO) production and iNOS expression by increasing HO-1 expression. It exhibits a potential as a nutritional preventive measure for neurodegenerative disorders. | ||
| S9871 | BTYNB | BTYNB (BTYNB IMP1 Inhibitor, MDK6620) is a potent and selective inhibitor of IMP1 binding to c-Myc mRNA. BTYNB downregulates β-TrCP1 mRNA and reduces activation of nuclear transcriptional factors-kappa B (NF-κB). BTYNB disrupts this enhancer function by impairing IGF2 mRNA-binding protein 1 (IGF2BP1)-RNA association. | ||
| S9106 | Eleutheroside E | Eleutheroside E, a principal component of Eleutherococcus senticosus (ES), has anti-inflammatory effects by inhibiting NF-κB and protecting against myocardial infarction. | ||
| S4460 | IMM-H007 | IMM-H007, an adenosine derivative, is an activator of AMP-Activated Protein Kinase (AMPK). IMM-H007 is a potential drug for treating cardiac dysfunction. IMM-H007 negatively regulates endothelium inflammation through inactivating NF-κB and JNK/AP1 signaling. IMM-H007 inhibits ABCA1 (ATP binding cassette subfamily a member 1) degradation and facilitates its cell-surface localization in macrophages, thereby promotes cholesterol efflux. | ||
| S5579 | Chelidonic acid | Chelidonic acid (Jerva acid, Jervaic acid, γ-Pyrone-2,6-dicarboxylic acid) is a secondary metabolite found in several plants with therapeutic potential in allergic disorders in experimental animals. Chelidonic acid inhibits IL-6 production by blocking NF-κB and caspase-1 in HMC-1 cells. Chelidonic acid is also an inhibitor of glutamate decarboxylase with Ki of 1.2 μM. | ||
| S8941 | NIK SMI1 | NIK SMI1 is a highly potent and selective NF-κB-inducing kinase (NIK) inhibitor with Ki of 0.23 nM for NIK-catalyzed hydrolysis of ATP to ADP. | ||
| E1798New | HOIPIN-8 | HOIPIN-8, is a potent inhibitor of linear ubiquitin chain assembly complex (LUBAC) and NF-κB signaling with an IC50 value of 11 nM. It serves as a powerful tool for investigating the physiological and cellular functions of LUBAC. | ||
| E0526 | sappanone A | Sappanone A, a homoisoflavanone isolated from the heartwood of Caesalpinia sappan (Leguminosae), induces HO-1 expression by activating Nrf2 through the p38 MAPK pathway, suppresses LPS-induced NF-κB activation by suppressing the phosphorylation of RelA/p65 at Ser536, exerting its anti-inflammatory effect. | ||
| S3138 | Methylthiouracil | Methylthiouracil (NSC-193526, NSC-9378,MTU) is an antithyroid agent. Methylthiouracil suppresses the production TNF-α and IL-6, and the activation of NF-κB and ERK1/2. | ||
| S3899 | Hederagenin | Hederagenin (Caulosapogenin, Hederagenol, Hederagenic acid, Astrantiagenin E) is a highly water insoluble triterpenoid compound that can be found in various plants including Hedera helix and Chenopodium quinoa. It exhibits a variety of biological activities, including potent antitumor properties both in vitro and in vivo. Hederagenin inhibits LPS-stimulated expression of iNOS, COX-2, and NF-κB. | ||
| E2661 | Chitosan oligosaccharide | Chitosan oligosaccharide (COS) is an oligomer of β-(1➔4)-linked d-glucosamine, of which actions involve the modulation of several important pathways including the suppression of nuclear factor kappa B (NF-κB) and mitogen-activated protein kinases (MAPK) and the activation of AMP-activated protein kinase (AMPK). | ||
| S1321 | Urolithin B | Urolithin B inhibits NF-κB activity by reducing the phosphorylation and degradation of IκBα. Urolithin B suppresses the phosphorylation of JNK, ERK, and Akt, and enhances the phosphorylation of AMPK. Urolithin B is also a regulator of skeletal muscle mass. | ||
| S3867 | (E)-Cardamonin | (E)-Cardamonin (Alpinetin chalcone, cardamomin) is a naturally occurring chalcone with strong anti-inflammatory activity. It is a novel TRPA1 antagonist with IC50 of 454 nM and also a NF-κB inhibitor. | ||
| S6714 | INH14 | INH14 is an inhibitor of TLR2-mediated NF-κB activation with IC50 values of 8.975 μM and 3.598 μM for IKKα and IKKβ, respectively. | ||
| E1714New | B022 | B022 is a potent and selective NF-κB-inducing kinase (NIK) inhibitor with a Ki of 4.2 nM. B022 protects liver from toxin-induced inflammation, oxidative stress, and injury. | ||
| S3261 | Myrislignan | Myrislignan, a lignan isolated from Myristica fragrans Houtt, possesses anti-inflammatory activities. Myrislignan inhibits interleukin-6 (IL-6) and tumour necrosis factor-α (TNF-α). Myrislignan significantly inhibits the expressions of inducible NO synthase (iNOS) and cyclooxygenase-2 (COX-2) dose-dependently in LPS-stimulated macrophage cells. Myrislignan inhibits the NF-κB signalling pathway activation. | ||
| E3786 | Radix Scrophulariae Extract | Radix Scrophulariae Extract is derived from Radix Scrophulariae, which has anti-apoptotic and anti-inflammatory effects, potentially operating by affecting the mitogen-activated protein kinases (MAPKs) signaling pathway and inhibition of the NF-κB pathway. | ||
| S3872 | Guaiacol | Guaiacol (O-methoxyphenol, 2-hydroxyanisole, O-methylcatechol) is a phenolic natural product first isolated from Guaiac resin and the oxidation of lignin. It is a precursor to various flavorants, such as eugenoland vanillin. Guaiacol, a phenolic compound isolated from Guaiac resin, inhibits LPS-stimulated COX-2 expression and NF-κB activation. Anti-inflammatory activity. | ||
| S4712 | Diethylmaleate | Diethylmaleate (Diethyl ester, Maleic acid diethyl ester) is the diethyl ester of maleic acid and a glutathione-depleting compound that inhibits NF-κB. | ||
| S3298 | Caulophylline (N-Methylcytisine) | Caulophylline (N-Methylcytisine, Caulophyllin, NMC) is a tricyclic quinolizidine alkaloid with anti-inflammatory activities. Caulophylline binds to nicotinic acetylcholine receptors (nAChR) from squid optical ganglia with Kd of 50 nM. Caulophylline significantly reduces myeloperoxidase (MPO) activity, blocks the activation of NF-κB by inhibiting IκB and IKK phosphorylation. | ||
| E3795 | Commelina Communis Extract | Commelina Communis Extract is drawed from Commelina Communis, which has anti-inflammatory effects related to suppression of the NF-κB pathway and can be used to treat chronic inflammation. | ||
| S3256 | Tectochrysin | Tectochrysin (Techtochrysin, NSC 80687) is one of the major flavonoids of Alpinia oxyphylla Miquel. Tectochrysin significantly increases the expression of DR3, DR4 and Fas and inhibits activity of NF-κB. Tectochrysin induces apoptotic cell death. | ||
| E0783 | SM-7368 | SM-7368 is a potent NF-κB inhibitor that targets downstream of MAPK p38 activation, also inhibits TNF-α-induced MMP-9 upregulation. | ||
| S3609 | Berbamine dihydrochloride | Berbamine (BA, BBM) dihydrochloride, a traditional Chinese medicines extracted from Berberis amurensis (xiaoboan), is a novel inhibitor of bcr/abl fusion gene with potent anti-leukemia activity and also an inhibitor of NF-κB. Berbamine (BA, BBM) dihydrochloride induces apoptosis in human myeloma cells and inhibits the growth of cancer cells by targeting Ca²⁺/calmodulin-dependent protein kinase II (CaMKII). | ||
| S3299 | Demethyleneberberine | Demethyleneberberine (DMB), a component of Cortex Phellodendri Chinensis (CPC), significantly alleviates the weight loss and diminishes myeloperoxidase (MPO) activity, while significantly reduces the production of pro-inflammatory cytokines, such as interleukin (IL)-6 and tumor necrosis factor-α (TNF-α), and inhibits the activation of NF-κB signaling pathway. Demethyleneberberine (DMB) potentially ameliorates NAFLD (Non-alcoholic fatty liver disease) by activating AMPK pathways. | ||
| A2020 | Denosumab (anti-RANK ligand) | Denosumab (anti-RANK ligand) (AMG-162) is a fully human monoclonal antibody that selectively inhibits primate RANKL. Denosumab inhibits the ability of human RANKL (143–317) to stimulate the formation of osteoclasts derived from murine RAW 264.7 cells with an IC50 value of 1.64 nM. | ||
| S3603 | Betulinic acid | Betulinic acid (ALS-357, Lupatic acid, Betulic acid), a pentacyclic triterpenoid from Syzigium claviflorum, is a inhibitor of HIV-1 with EC50 of 1.4 μ M. It's reported that Betulinic acid acts as a new activator of NF-κB.Phase 1/2. | ||
| S2979 | CU-T12-9 | CU-T12-9 is a specific agonist of Toll-like receptor (TLR) 1/2 with EC50 of 52.9 nM in HEK-Blue hTLR2 SEAP assay. CU-T12-9 activates both the innate and the adaptive immune systems. CU-T12-9 signals through NF-κB and invokes an elevation of the downstream effectors TNF-α, IL-10, and iNOS. | ||
| E1351 | NF-κΒ activator 1 | NF-κΒ activator 1(Compound 32) is a potent NF-κΒ activator, with an EC50 of 0.9 μM. | ||
| S5804 | N-Acetylcysteine amide | N-acetylcysteine amide is a membrane penetrating antioxidant with anti-inflamatory activity through regulation of activation of NF-κB and HIF-1α as well as modulation of ROS. | ||
| S4611 | TCEP Hydrochloride | TCEP (Tris(2-carboxyethyl)phosphine) hydrochloride, a non-thiol reducing agent, promotes NF-κB-DNA binding in a dose-related manner.Solutions are best fresh-prepared. | ||
| E3566 | Largeleaf Gentian Root Extract | Largeleaf Gentian Root Extract is extracted from the root of Gentiana macrophylla, which can modulate the CD147/p38/NF-κB pathway. | ||
| S0949 | Cucurbitacin IIb | Cucurbitacin IIb (CuIIb, Dihydrocucurbitacin F, 25-deacetyl hemslecin A) inhibits phosphorylation of STAT3, JNK and Erk1/2, enhances the phosphorylation of IκB and NF-κB, blocks nuclear translocation of NF-κB and decreases mRNA levels of IκBα and TNF-α. Cucurbitacin IIb exhibits anti-inflammatory activity and induces apoptosis. Cucurbitacin IIb is isolated from Hemsleya amabilis. | ||
| E3625 | Cnidii Fructus Extract | Cnidii Fructus Extract is extracted from Cnidii Fructus, the dry ripe fruit of Cnidium monnieri (L.)Cuss.. Chinese herbal medicine Cnidii Fructus has an outstanding effect on chronic lumbar pain and impotence, also has been used against osteoporosis with high frequency. | ||
| E3594 | Radix tetrastigme Extract | Radix Tetrastigme Extract is extracted from Tetrastigma hemsleyanum, which can modulate TLR4/COX-2/NF-κB signaling pathway. | ||
| S9184 | Forsythoside B | Forsythoside B is a phenylethanoid glycoside isolated from the leaves of Lamiophlomis rotata Kudo, a Chinese folk medicinal plant. Forsythoside B has potent neuroprotective effects and has anti-inflammation, antioxidant, and antisepsis properties. Forsythoside B could inhibit TNF-alpha, IL-6, IκB and modulate NF-κB. | ||
| E0211 | Dihydroberberine | Dihydroberberine, a hydrogenated derivative of Berberine (BBR), exerts anti-inflammatory effect via dual modulation of NF-κB and MAPK signaling pathways. |
||
| E4443New | S-Nitrosoglutathione | S-nitrosoglutathione (GSNO, RVC-588, S-Nitroso-L-glutathione,Nitrosoglutathione) is an amino acid and nitric oxide donor that, under physiological conditions, spontaneously releases nitric oxide. S-nitrosoglutathione (GSNO) selectively inhibits platelet and NF-κB activation. It induces apoptosis in T cells in low concentrations (< 0.6 mM) while protecting T cells from apoptosis in higher concentrations (1-2 mM). | ||
| P1238New | SN 52 | SN52, a cell-permeable peptide containing the p50 NLS, is a selective inhibitor of NF-κB pathway. It selectively and completely blocks the ionizing radiation (IR)-induced nuclear import of p52 and RelB. It also enhances the radiosensitivity of prostate cancer cells. | ||
| E1575New | EN450 | EN450 is a cysteine-reactive covalent molecular glue degrader targeting NF-κB. It interacts with allosteric C111 in the E2 ubiquitin ligase UBE2D that induces the ternary complex formation between UBE2D and NFKB1. It exhibits anti-proliferative effects through a Cullin E3 ligase and proteasome-dependent mechanism. | ||
| E1798New | HOIPIN-8 | HOIPIN-8, is a potent inhibitor of linear ubiquitin chain assembly complex (LUBAC) and NF-κB signaling with an IC50 value of 11 nM. It serves as a powerful tool for investigating the physiological and cellular functions of LUBAC. | ||
| E1714New | B022 | B022 is a potent and selective NF-κB-inducing kinase (NIK) inhibitor with a Ki of 4.2 nM. B022 protects liver from toxin-induced inflammation, oxidative stress, and injury. |
Choose Selective NF-κB Inhibitors
NF-κBSignaling Pathway
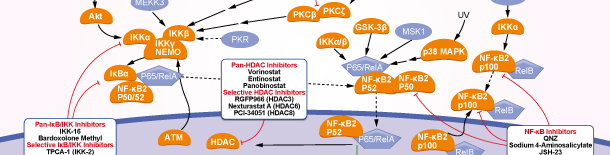
Tags: NF-κB inhibitor|NF-κB agonist|NF-κB activator|NF-κB inducer|NF-κB antagonist|NF-κB signaling pathway|NF-κB assay kit
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.