-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
research use only
Mocetinostat (MGCD0103) HDAC inhibitor
Cat.No.S1122
Chemical Structure
Molecular Weight: 396.44
Quality Control
Cell Culture, Treatment & Working Concentration
| Cell Lines | Assay Type | Concentration | Incubation Time | Formulation | Activity Description | PMID |
|---|---|---|---|---|---|---|
| PBMC | Apoptosis Assay | 0.5/2/3 μM | 24/48 h | induces apoptosis dose and time dependently | 20406947 | |
| HeLa | Function Assay | 10 μM | 7 h | DMSO | disrupts normal spindle checkpoint function | 20538840 |
| HeLa | Function Assay | 10 μM | 6/12/24 h | DMSO | induces mitotic accumulation and delayed p21 expression | 20538840 |
| HeLa | Function Assay | 0.3-10 μM | 8 h | DMSO | increases caspase 3 and 7 activation dose dependently | 20538840 |
| HeLa | Function Assay | 0.3-10 μM | 8 h | DMSO | increases acetylated H3 K9 (H3K9Ac) at 10 μM | 20538840 |
| DMS114 | Growth Inhibition Assay | IC50=640 nM | 20682643 | |||
| H82 | Growth Inhibition Assay | IC50=250 nM | 20682643 | |||
| H146 | Growth Inhibition Assay | IC50=35 nM | 20682643 | |||
| H526 | Growth Inhibition Assay | IC50=480 nM | 20682643 | |||
| KM-H2 | Function Assay | 1 μM | 0.25-48 h | DMSO | activates NF-kB | 20880107 |
| L428 | Function Assay | 1 μM | 0.25-48 h | DMSO | activates NF-kB | 20880107 |
| HD-LM2 | Function Assay | 1 μM | 0.25-48 h | DMSO | activates NF-kB | 20880107 |
| KM-H2 | Function Assay | 0.5/1 μM | 24/48 h | DMSO | upregulates TNF-α dose and time dependently | 20880107 |
| L428 | Function Assay | 0.5/1 μM | 24/48 h | DMSO | upregulates TNF-α dose and time dependently | 20880107 |
| HD-LM2 | Function Assay | 0.5/1 μM | 24/48 h | DMSO | upregulates TNF-α dose and time dependently | 20880107 |
| KM-H2 | Function Assay | 1 μM | 24/48 h | DMSO | downregulates XIAP, activated caspases 9 and 3 | 20880107 |
| L428 | Function Assay | 1 μM | 24/48 h | DMSO | downregulates XIAP, activated caspases 9 and 3 | 20880107 |
| HD-LM2 | Function Assay | 1 μM | 24/48 h | DMSO | downregulates XIAP, activated caspases 9 and 3 | 20880107 |
| KM-H2 | Apoptosis Assay | 0.1/0.5/1 μM | 48 h | DMSO | induces apoptosis dose dependently | 20880107 |
| L428 | Apoptosis Assay | 0.1/0.5/1 μM | 48 h | DMSO | induces apoptosis dose dependently | 20880107 |
| HD-LM2 | Apoptosis Assay | 0.1/0.5/1 μM | 48 h | DMSO | induces apoptosis dose dependently | 20880107 |
| KM-H2 | Function Assay | 0.1-2 μM | 24 h | DMSO | shows acetylation of histone 3 and upregulation of the cell cycle regulatory protein p21 | 20880107 |
| L428 | Function Assay | 0.1-2 μM | 24 h | DMSO | shows acetylation of histone 3 and upregulation of the cell cycle regulatory protein p21 | 20880107 |
| HD-LM2 | Function Assay | 0.1-2 μM | 24 h | DMSO | shows acetylation of histone 3 and upregulation of the cell cycle regulatory protein p21 | 20880107 |
| KM-H2 | Growth Inhibition Assay | 72 h | DMSO | IC50=2.86 μM | 20880107 | |
| L428 | Growth Inhibition Assay | 72 h | DMSO | IC50=1.96 μM | 20880107 | |
| HD-LM2 | Growth Inhibition Assay | 72 h | DMSO | IC50=1.88 μM | 20880107 | |
| LP1 | Function Assay | 1 μM | 24 h | enhances 5-AC-induced MAGE-A3 gene expression | 21171821 | |
| ANBL6 | Function Assay | 1 μM | 24 h | enhances 5-AC-induced MAGE-A3 gene expression | 21171821 | |
| HMEC | Growth Inhibition Assay | IC50=19 μM | 21317455 | |||
| SW620 | Growth Inhibition Assay | IC50=1 μM | 21317455 | |||
| SW48 | Growth Inhibition Assay | IC50=0.8 μM | 21317455 | |||
| HT-29 | Growth Inhibition Assay | IC50=0.7 μM | 21317455 | |||
| HCT15 | Growth Inhibition Assay | IC50=0.7 μM | 21317455 | |||
| PAXF 1657L† | Growth Inhibition Assay | EC50=0.3 μM | 21375679 | |||
| PAXF 546L† | Growth Inhibition Assay | EC50=1.5 μM | 21375679 | |||
| Panc-1 | Growth Inhibition Assay | EC50=1.8 μM | 21375679 | |||
| MiaPaca-2 | Growth Inhibition Assay | EC50=0.6 μM | 21375679 | |||
| AsPC-1 | Growth Inhibition Assay | EC50=3.9 μM | 21375679 | |||
| BxPC-3 | Growth Inhibition Assay | EC50=1.1 μM | 21375679 | |||
| MMCs | Function Assay | 1 μM | 6-24 h | dose-dependently inhibits the trimethylation level of H3-K9 (H3-K9me3) | 24451378 | |
| MMCs | Function Assay | 1 μM | 24 h | augments global acetylation levels of histone H3-K9/14 (H3-K9/14ac) and H4-K12 (H4-K12ac) | 24451378 | |
| MMCs | Function Assay | 1 μM | 24 h | increases HAT activity | 24451378 | |
| MMCs | Function Assay | 0.5/1 μM | 24 h | shows 45-fold stimulation in cGMP levels | 24451378 | |
| MMCs | Function Assay | 1 μm | 0-48 h | increases NPRA protein expression 2.7–3.5 fold | 24451378 | |
| Panc1 | Cell Viability Assay | 1 μM | 72 h | DMSO | enhances gemcitabine-induces cell viability decrease | 25872941 |
| Panc1 | Apoptosis Assay | 1 μM | 72 h | DMSO | sensitizes Panc1 cells for gemcitabine-induced apoptosis | 25872941 |
| Panc1 | Function Assay | 0.5/1/2.5 μM | 48 h | DMSO | reduces expression of ZEB1 on both mRNA and protein level | 25872941 |
| Panc1 | Function Assay | 0.5/1/2.5 μM | 48 h | DMSO | upregulates miR-203 | 25872941 |
| MOLP8 | Growth Inhibition Assay | 48 h | IC50=0.6± 0.04μM | 26091518 | ||
| T47D | Growth Inhibition Assay | 48 h | IC50=1.17 μM | 26378038 | ||
| MCF7 | Growth Inhibition Assay | 48 h | IC50=0.67 μM | 26378038 | ||
| BT549 | Growth Inhibition Assay | 48 h | IC50=4.38 μM | 26378038 | ||
| MDA-MB-231 | Growth Inhibition Assay | 48 h | IC50=3.04 μM | 26378038 | ||
| HEK293 | Function assay | Inhibition of HDAC1 in HEK293 cells, IC50=0.13μM | 18308563 | |||
| HEK293 | Function assay | Inhibition of HDAC3 in HEK293 cells, IC50=0.61μM | 18308563 | |||
| HCT116 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human HCT116 cells after 72 hrs by MTT assay, IC50=0.29μM | 18570366 | ||
| HCT116 | Function assay | Induction of p21cip/waf1 protein expression in human HCT116 cells relative to MS275, EC50=0.45μM | 18570366 | |||
| Du145 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human Du145 cells after 72 hrs by MTT assay, IC50=0.67μM | 18570366 | ||
| A549 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human A549 cells after 72 hrs by MTT assay, IC50=0.9μM | 18570366 | ||
| HCT116 | Cell cycle assay | Cell cycle arrest in human HCT116 cells assessed as accumulation at G2/M phase, EC50<1μM | 18570366 | |||
| T24 | Function assay | Induction of H3 histone acetylation in human T24 cells relative to MS275, EC50=1.38μM | 18570366 | |||
| HCT116 | Apoptosis assay | 1 uM | Induction of apoptosis in HCT116 cells at 1 uM | 18570366 | ||
| HCT116 | Antiproliferative assay | Antiproliferative activity against human HCT116 cells by MTT assay, IC50=0.3μM | 19114304 | |||
| HCT116 | Function assay | 16 hrs | Induction of p21WAF1/CIP1 expression in human HCT116 cells assessed as tubulin level after 16 hrs by luciferase assay, EC50=0.6μM | 19114304 | ||
| T24 | Function assay | 16 hrs | Induction of histone H4 hyperacetylation in human T24 cells after 16 hrs by immunoblotting, EC50<1μM | 19114304 | ||
| HCT116 | Antiproliferative assay | Antiproliferative activity against human HCT116 cells assessed as growth inhibition, IC50=0.31μM | 21650221 | |||
| H1299 | Antiproliferative assay | Antiproliferative activity against human H1299 cells, IC50=1.44μM | 21650221 | |||
| HCT116 | Antiproliferative assay | Antiproliferative activity against human HCT116 cells, IC50=0.31μM | 21742496 | |||
| Sf9 | Function assay | 2 hrs | Inhibition of human recombinant HDAC1 expressed in Sf9 cells incubated for 2 hrs using RHKK-Ac fluorogenic substrate, IC50=0.102μM | 23009203 | ||
| HCT116 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HCT116 cells after 72 hrs by MTT assay, IC50=0.327μM | 23206867 | ||
| A549 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human A549 cells after 72 hrs by MTT assay, IC50=1.279μM | 23206867 | ||
| MCF7 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MCF7 cells after 72 hrs by MTT assay, IC50=4.807μM | 23206867 | ||
| HCT116 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HCT116 cells assessed as growth inhibition by measuring cellular ATP level after 72 hrs by cell-titer glo assay, IC50=0.7μM | 23829483 | ||
| MCF7 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MCF7 cells assessed as growth inhibition by measuring cellular ATP level after 72 hrs by cell-titer glo assay, IC50=1.26μM | 23829483 | ||
| A549 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human A549 cells assessed as growth inhibition by measuring cellular ATP level after 72 hrs by cell-titer glo assay, IC50=1.73μM | 23829483 | ||
| DU145 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human DU145 cells assessed as growth inhibition by measuring cellular ATP level after 72 hrs by cell-titer glo assay, IC50=2.06μM | 23829483 | ||
| Jurkat | Apoptosis assay | 1 to 10 uM | 24 hrs | Induction of apoptosis in human Jurkat cells assessed as PARP cleavage at 1 to 10 uM after 24 hrs by Western blot analysis | 23829483 | |
| HeLa | Function assay | 1 to 10 uM | 24 hrs | Inhibition of HDAC in human HeLa cells assessed as increase in H3K9Ac level at 1 to 10 uM after 24 hrs by Western blot analysis | 23829483 | |
| Jurkat | Function assay | 1 to 10 uM | 24 hrs | Inhibition of HDAC in human Jurkat cells assessed as increase in H3K9Ac level at 1 to 10 uM after 24 hrs by Western blot analysis | 23829483 | |
| High5 | Function assay | 3 to 24 hrs | Inhibition of human recombinant HDAC1 expressed in baculovirus infected insect high5 cells using Ac-Lys-Tyr-Lys (epsilon-acetyl)-AMC as substrate after 3 to 24 hrs by fluorescence assay, IC50=0.95μM | 24095018 | ||
| HCT116 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human HCT116 cells assessed as cell viability after 72 hrs by MTT assay, IC50=1.57μM | 24095018 | ||
| A549 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human A549 cells assessed as cell viability after 72 hrs by MTT assay, IC50=1.65μM | 24095018 | ||
| High5 | Function assay | 3 to 24 hrs | Inhibition of human recombinant HDAC3 expressed in baculovirus infected insect high5 cells using Ac-Lys-Tyr-Lys (epsilon-acetyl)-AMC as substrate after 3 to 24 hrs by fluorescence assay, IC50=1.67μM | 24095018 | ||
| SNU16 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human SNU16 cells assessed as growth inhibition after 72 hrs by MTT assay, IC50=0.142μM | 25805446 | ||
| High5 | Function assay | 24 hrs | Inhibition of recombinant human HDAC2 expressed in baculovirus infected insect High5 cells using Ac-Lys-Tyr-Lys (epsilon-acetyl)-AMC as substrate after 24 hrs by fluorescence assay, IC50=0.17μM | 25805446 | ||
| High5 | Function assay | 3 hrs | Inhibition of recombinant human HDAC3 expressed in baculovirus infected insect High5 cells using Ac-Lys-Tyr-Lys (epsilon-acetyl)-AMC as substrate after 3 hrs by fluorescence assay, IC50=0.36μM | 25805446 | ||
| High5 | Function assay | 24 hrs | Inhibition of recombinant human HDAC1 expressed in baculovirus infected insect High5 cells using Ac-Lys-Tyr-Lys (epsilon-acetyl)-AMC as substrate after 24 hrs by fluorescence assay, IC50=0.39μM | 25805446 | ||
| HCT116 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HCT116 cells assessed as growth inhibition after 72 hrs by MTT assay, IC50=0.396μM | 25805446 | ||
| SW620 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human SW620 cells assessed as growth inhibition after 72 hrs by MTT assay, IC50=0.419μM | 25805446 | ||
| MKN45 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MKN45 cells assessed as growth inhibition after 72 hrs by MTT assay, IC50=0.61μM | 25805446 | ||
| Hep3B | Cytotoxicity assay | 72 hrs | Cytotoxicity against human Hep3B cells assessed as growth inhibition after 72 hrs by MTT assay, IC50=0.823μM | 25805446 | ||
| HepG2 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HepG2 cells assessed as growth inhibition after 72 hrs by MTT assay, IC50=0.876μM | 25805446 | ||
| SNU5 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human SNU5 cells assessed as growth inhibition after 72 hrs by MTT assay, IC50=1.009μM | 25805446 | ||
| A549 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human A549 cells assessed as growth inhibition after 72 hrs by MTT assay, IC50=2.08μM | 25805446 | ||
| SJSA1 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human SJSA1 cells assessed as growth inhibition after 72 hrs by MTT assay, IC50=3.624μM | 25805446 | ||
| MHCC97H | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MHCC97H cells assessed as growth inhibition after 72 hrs by MTT assay, IC50=4.563μM | 25805446 | ||
| PANC1 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human PANC1 cells assessed as growth inhibition after 72 hrs by MTT assay, IC50=26.774μM | 25805446 | ||
| U937 | Function assay | 10 uM | 24 hrs | Inhibition of HDAC3 in human U937 cells assessed as increase in histone H3 lysine-9 acetylation at 10 uM incubated for 24 hrs by Western blotting method | 26287310 | |
| PC3 | Function assay | 10 uM | 24 hrs | Inhibition of HDAC3 in human PC3 cells assessed as increase in histone H3 lysine-9 acetylation at 10 uM incubated for 24 hrs by Western blotting method | 26287310 | |
| U937 | Function assay | 10 uM | 24 hrs | Inhibition of HDAC in human U937 cells assessed as reduction in cyclin E expression in at 10 uM incubated for 24 hrs by Western blotting method | 26287310 | |
| Sf9 | Function assay | 10 mins | Inhibition of recombinant full length human C-terminal FLAG-tagged HDAC11 expressed in baculovirus infected Sf9 cells using Boc-Lys(epsilon-Ac)-AMC as substrate pretreated for 10 mins followed by substrate addition by fluorometric method, IC50=0.59μM | 28501514 | ||
| TC32 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for TC32 cells | 29435139 | |||
| DAOY | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for DAOY cells | 29435139 | |||
| SJ-GBM2 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SJ-GBM2 cells | 29435139 | |||
| A673 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for A673 cells | 29435139 | |||
| SK-N-MC | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SK-N-MC cells | 29435139 | |||
| BT-37 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for BT-37 cells | 29435139 | |||
| NB-EBc1 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for NB-EBc1 cells | 29435139 | |||
| U-2 OS | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for U-2 OS cells | 29435139 | |||
| Saos-2 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for Saos-2 cells | 29435139 | |||
| SK-N-SH | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SK-N-SH cells | 29435139 | |||
| NB1643 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for NB1643 cells | 29435139 | |||
| LAN-5 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for LAN-5 cells | 29435139 | |||
| BT-12 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for BT-12 cells | 29435139 | |||
| Rh18 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for Rh18 cells | 29435139 | |||
| OHS-50 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for OHS-50 cells | 29435139 | |||
| RD | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for RD cells | 29435139 | |||
| MG 63 (6-TG R) | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for MG 63 (6-TG R) cells | 29435139 | |||
| fibroblast cells | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for control Hh wild type fibroblast cells | 29435139 | |||
| Rh41 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for Rh41 cells | 29435139 | |||
| A673 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for A673 cells) | 29435139 | |||
| DAOY | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for DAOY cells | 29435139 | |||
| SJ-GBM2 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for SJ-GBM2 cells | 29435139 | |||
| U-2 OS | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for U-2 OS cells | 29435139 | |||
| RD | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for RD cells | 29435139 | |||
| Rh18 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for Rh18 cells | 29435139 | |||
| Rh30 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for Rh30 cells | 29435139 | |||
| Sf9 | Function assay | Inhibition Assay: HDAC inhibition assays were performed by Reaction Biology Corp. (Malvern, Pa.) using isolated human, recombinant full-length HDAC1 and -6 from a baculovirus expression system in Sf9 cells, IC50=0.102μM | ChEMBL | |||
| HCT116 | Antiproliferative assay | 48 hrs | Antiproliferative activity against human HCT116 cells assessed as reduction in cell viability after 48 hrs by MTT assay, IC50=1.24μM | ChEMBL | ||
| MCF7 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human MCF7 cells assessed as reduction in cell viability after 72 hrs by MTT assay, IC50=2.49μM | ChEMBL | ||
| HeLa | Antiproliferative assay | 48 hrs | Antiproliferative activity against human HeLa cells assessed as reduction in cell viability after 48 hrs by MTT assay, IC50=3.32μM | ChEMBL | ||
| HeLa | Antiproliferative assay | 72 hrs | Antiproliferative activity against human HeLa cells assessed as reduction in cell viability after 72 hrs by MTT assay, IC50=3.42μM | ChEMBL | ||
| HCT116 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human HCT116 cells assessed as reduction in cell viability after 72 hrs by MTT assay, IC50=3.51μM | ChEMBL | ||
| HepG2 | Antiproliferative assay | 48 hrs | Antiproliferative activity against human HepG2 cells assessed as reduction in cell viability after 48 hrs by MTT assay, IC50=4.05μM | ChEMBL | ||
| HepG2 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human HepG2 cells assessed as reduction in cell viability after 72 hrs by MTT assay, IC50=4.25μM | ChEMBL | ||
| HepG2 | Antiproliferative assay | 24 hrs | Antiproliferative activity against human HepG2 cells assessed as reduction in cell viability after 24 hrs by MTT assay, IC50=5.79μM | ChEMBL | ||
| A549 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human A549 cells assessed as reduction in cell viability after 72 hrs by MTT assay, IC50=11.87μM | ChEMBL | ||
| A549 | Antiproliferative assay | 48 hrs | Antiproliferative activity against human A549 cells assessed as reduction in cell viability after 48 hrs by MTT assay, IC50=14.57μM | ChEMBL | ||
| HCT116 | Antiproliferative assay | 24 hrs | Antiproliferative activity against human HCT116 cells assessed as reduction in cell viability after 24 hrs by MTT assay, IC50=29.69μM | ChEMBL | ||
| HeLa | Antiproliferative assay | 24 hrs | Antiproliferative activity against human HeLa cells assessed as reduction in cell viability after 24 hrs by MTT assay, IC50=43.8μM | ChEMBL | ||
| Click to View More Cell Line Experimental Data | ||||||
Solubility
|
In vitro |
DMSO
: 60 mg/mL
(151.34 mM)
Water : Insoluble Ethanol : Insoluble |
Molarity Calculator
|
In vivo |
|||||
In vivo Formulation Calculator (Clear solution)
Step 1: Enter information below (Recommended: An additional animal making an allowance for loss during the experiment)
Step 2: Enter the in vivo formulation (This is only the calculator, not formulation. Please contact us first if there is no in vivo formulation at the solubility Section.)
Calculation results:
Working concentration: mg/ml;
Method for preparing DMSO master liquid: mg drug pre-dissolved in μL DMSO ( Master liquid concentration mg/mL, Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug. )
Method for preparing in vivo formulation: Take μL DMSO master liquid, next addμL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O, mix and clarify.
Method for preparing in vivo formulation: Take μL DMSO master liquid, next add μL Corn oil, mix and clarify.
Note: 1. Please make sure the liquid is clear before adding the next solvent.
2. Be sure to add the solvent(s) in order. You must ensure that the solution obtained, in the previous addition, is a clear solution before proceeding to add the next solvent. Physical methods such
as vortex, ultrasound or hot water bath can be used to aid dissolving.
Chemical Information, Storage & Stability
| Molecular Weight | 396.44 | Formula | C23H20N6O |
Storage (From the date of receipt) | |
|---|---|---|---|---|---|
| CAS No. | 726169-73-9 | Download SDF | Storage of Stock Solutions |
|
|
| Synonyms | MG0103 | Smiles | C1=CC=C(C(=C1)N)NC(=O)C2=CC=C(C=C2)CNC3=NC=CC(=N3)C4=CN=CC=C4 | ||
Mechanism of Action
| Targets/IC50/Ki |
HDAC1
(Cell-free assay) 0.15 μM
HDAC2
(Cell-free assay) 0.29 μM
HDAC11
(Cell-free assay) 0.59 μM
HDAC3
(Cell-free assay) 1.66 μM
|
|---|---|
| In vitro |
Mocetinostat (MGCD0103) inhibits only a subset of the nine human recombinant HDACs, including HDAC1, HDAC2, HDAC3, and HDAC11 at nanomolar or low micromolar concentrations, in a dose-dependent manner. It reveals most potent inhibitory activity against human HDAC1 and HDAC2 enzymes in vitro, and does not inhibit class II HDACs. The exocyclic amino group in this compound is necessary for enzyme inhibitory activity because HDAC-inhibitory activity against HDAC1 and HDAC2 is completely abolished with the desamino analogue. Its inhibitory activity reaches the maximum plateau at 6 μM, and the maximal inhibitable enzyme pool affected by MGCD0103 is 75% of the total enzyme activity in HCT116 cells whereas NVP-LAQ824 inhibits almost 100% of that in these cells. In A549 cells, it also exhibits dose-dependent inhibition of HDAC activity in whole cells. |
| Kinase Assay |
HDAC enzyme assay in vitro
|
|
The deacetylase enzyme assay is based on a homogeneous fluorescence release assay. Purified recombinant HDAC enzymes are incubated with Mocetinostat (MGCD0103) diluted in various concentrations for 10 minutes in assay buffer [25 mM HEPES (pH 8.0), 137 mM NaCl, 1 mM MgCl2, 2.7 mM KCl] at room temperature. The substrate Boc-Lys(ε-Ac)-AMC is added to the reaction for further incubation at 37 °C. The concentration of the substrate and the incubation time varies for different isotypes of HDAC enzymes. A 20-min trypsin incubation at room temperature allows the release of the fluorophore from the deacetylated substrate. The fluorescent signal is detected by fluorometer at excitation of 360 nm, emission of 470 nm, and cutoff at 435 nm.
|
|
| In vivo |
Mocetinostat (MGCD0103) significantly inhibites growth of human tumor xenografts in nude mice and the antitumor activity correlated with induction of histone acetylation in tumors. P.O. administration of this compound (2HBr salt) significantly decreases growth of implanted advanced A549 tumors in nude mice in a dose-dependent manner after 13 days of daily administration. It (170 mg/kg for 2HBr salt, corresponding to 120 mg/kg of free base) significantly blockes growth of tumors compared with vehicle treatment alone with no change in body weight. In addition, it does not reduce WBC counts and is well tolerated. The compound is also orally active in many other human tumor xenograft models including NSCLC H1437. At 80 mg/kg (free base), it almost completely blocks the growth of H1437 tumors after 13 days of daily p.o. administration with no reduction of body weight in animals. It reduces pulmonary arterial pressure more dramatically. Moreover, this compound improves pulmonary artery acceleration time and reduced systolic notching of the pulmonary artery flow envelope, which suggests a positive impact of the HDAC inhibitor on pulmonary vascular remodeling and stiffening. |
References |
|
Applications
| Methods | Biomarkers | Images | PMID |
|---|---|---|---|
| Western blot | Ac-H3 / Ac-H4 / Ac-tubulin Bad / Bid / Bak / Puma / Bax / Cleaved caspase-9 / Cleaved caspase-3 / Cleaved PARP |
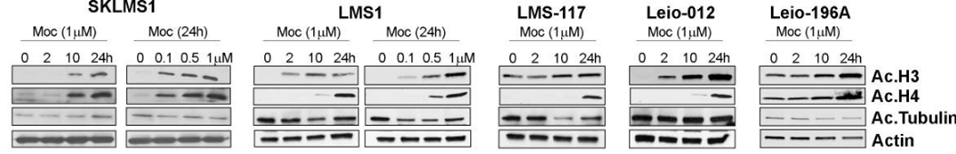
|
29186204 |
| Immunofluorescence | Nanog / MHC E-cadherin / ZEB1 |
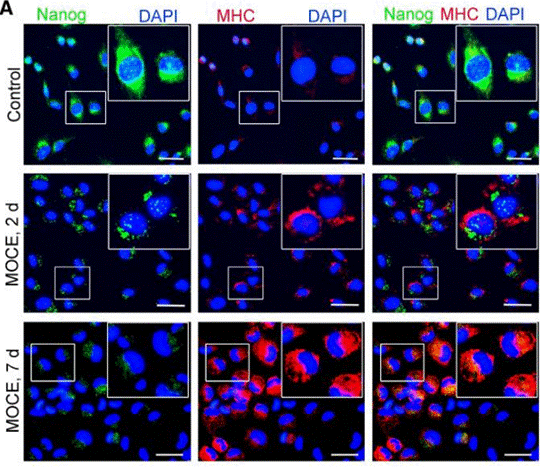
|
26240433 |
| Growth inhibition assay | Cell viability |
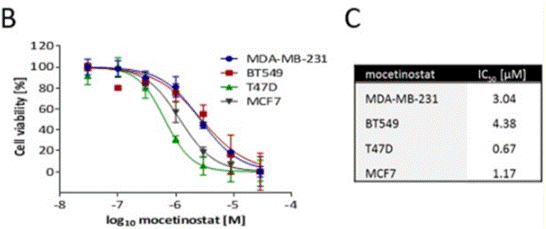
|
26378038 |
Clinical Trial Information
(data from https://clinicaltrials.gov, updated on 2024-05-22)
| NCT Number | Recruitment | Conditions | Sponsor/Collaborators | Start Date | Phases |
|---|---|---|---|---|---|
| NCT04299113 | Recruiting | Rhabdomyosarcoma |
Jonsson Comprehensive Cancer Center|Mirati Therapeutics Inc.|Phase One Foundation |
May 14 2020 | Phase 1 |
| NCT02993991 | Withdrawn | Squamous Cell Carcinoma Head And Neck|Squamous Cell Carcinoma Mouth|Resectable Squamous Cell Carcinoma of Oral Cavity |
University Health Network Toronto|Mirati Therapeutics Inc.|AstraZeneca |
October 10 2017 | Phase 1 |
| NCT02236195 | Completed | Urothelial Carcinoma |
Mirati Therapeutics Inc. |
October 2014 | Phase 2 |
| NCT00666497 | Terminated | Acute Myeloid Leukemia (AML)|Myelodysplastic Syndrome (MDS) |
Mirati Therapeutics Inc. |
June 2008 | Phase 2 |
| NCT00511576 | Terminated | Breast Cancer|Lung Cancer|Pulmonary Cancer|Non-Small-Cell Lung Carcinoma|Prostate Cancer|Prostatic Cancer|Gastric Cancer|Stomach Cancer |
Mirati Therapeutics Inc. |
August 2007 | Phase 1 |
Tech Support
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
Signaling Pathway Map
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.