-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
research use only
Milciclib CDK inhibitor
Cat.No.S2751
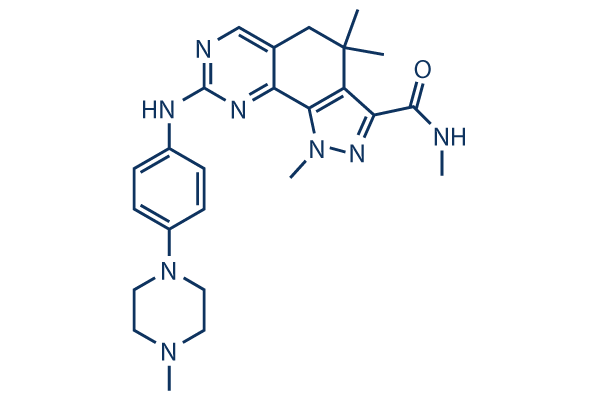
Chemical Structure
Molecular Weight: 460.57
Quality Control
Cell Culture, Treatment & Working Concentration
| Cell Lines | Assay Type | Concentration | Incubation Time | Formulation | Activity Description | PMID |
|---|---|---|---|---|---|---|
| human A2780 cells | Proliferation assay | 72 h | Antiproliferative activity against human A2780 cells after 72 hrs by cell Titer_Glo assay, IC50=0.2 μM | |||
| human A2780 cells | Function assay | Inhibition of CDK2 in human A2780 cells assessed as reduction of hyperphosphorylated form of retinoblastoma protein at 1 uM relative to control | ||||
| Click to View More Cell Line Experimental Data | ||||||
Solubility
|
In vitro |
DMSO
: 17 mg/mL
(36.91 mM)
Water : Insoluble Ethanol : Insoluble |
Molarity Calculator
|
In vivo |
|||||
In vivo Formulation Calculator (Clear solution)
Step 1: Enter information below (Recommended: An additional animal making an allowance for loss during the experiment)
Step 2: Enter the in vivo formulation (This is only the calculator, not formulation. Please contact us first if there is no in vivo formulation at the solubility Section.)
Calculation results:
Working concentration: mg/ml;
Method for preparing DMSO master liquid: mg drug pre-dissolved in μL DMSO ( Master liquid concentration mg/mL, Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug. )
Method for preparing in vivo formulation: Take μL DMSO master liquid, next addμL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O, mix and clarify.
Method for preparing in vivo formulation: Take μL DMSO master liquid, next add μL Corn oil, mix and clarify.
Note: 1. Please make sure the liquid is clear before adding the next solvent.
2. Be sure to add the solvent(s) in order. You must ensure that the solution obtained, in the previous addition, is a clear solution before proceeding to add the next solvent. Physical methods such
as vortex, ultrasound or hot water bath can be used to aid dissolving.
Chemical Information, Storage & Stability
| Molecular Weight | 460.57 | Formula | C25H32N8O |
Storage (From the date of receipt) | |
|---|---|---|---|---|---|
| CAS No. | 802539-81-7 | Download SDF | Storage of Stock Solutions |
|
|
| Synonyms | PHA-848125 | Smiles | CC1(CC2=CN=C(N=C2C3=C1C(=NN3C)C(=O)NC)NC4=CC=C(C=C4)N5CCN(CC5)C)C | ||
Mechanism of Action
| Targets/IC50/Ki |
CDK2/CyclinA
(Cell-free assay) 45 nM
TrkA
(Cell-free assay) 53 nM
CDK7/CyclinH
(Cell-free assay) 150 nM
CDK4/CyclinD1
(Cell-free assay) 160 nM
CDK5/p35
(Cell-free assay) 265 nM
CDK2/CyclinE
(Cell-free assay) 363 nM
CDK1/CyclinB
(Cell-free assay) 398 nM
|
|---|---|
| In vitro |
PHA-848125 inhibits, although with lower potency, the activities of cyclin H/CDK7, cyclin D1/CDK4, p35/CDK5, as well as cyclin E/CDK2 and cyclin B/CDK1 with IC50 values of 0.15, 0.16, 0.265, 0.363, 0.398 μM, respectively. Thropomyosin receptor kinase A is also inhibited by this compound in the same nanomolar range as CDKs. In the most PHA-848125-sensitive cell line, this compound induces a concentration-dependent G(1) arrest. This compound also impairs phosphorylation of the retinoblastoma protein at CDK2 and CDK4 specific sites, reduces retinoblastoma protein and cyclin A levels, and increases p21(Cip1), p27(Kip1) and p53 expression. This chemical is added to the cells 48 h after TMZ and cell growth is evaluated after 3 additional days of culture. A drug combination of TMZ, BG and this compound induces an additive or synergistic effect on cell growth, depending on the cell line. In the absence of BG, the combination is still more active than the single agents in cell lines moderately sensitive to TMZ, but comparable to this compound alone in the two TMZ-resistant cell lines. When TMZ plus BG are used in combination with this chemical against cultured normal melanocytes, neither synergistic nor additive antiproliferative effects are observed.
|
| Kinase Assay |
Biochemical kinase inhibition assays
|
|
Inhibition of kinase activity by this compound is assessed using a strong anion exchanger (Dowex 1X8 resin)–based assay in robotized format run on 384-well plates. In this assay, specific peptides or protein substrates are transphosphorylated by their specific kinase in the presence of ATP traced with [γ-33P]ATP using optimal buffers and cofactors. The potency of this chemical toward CDKs and 38 additional kinases belonging to an in-house Kinase Selectivity Screening panel is evaluated, and the relevant IC50s are determined. For each enzyme, the absolute KM values for ATP and the specific substrate are calculated and each assay is then run at optimized ATP (2KM) and substrate (5KM) concentrations. This setting enables direct comparison of IC50 values of this compound across the panel for evaluation of its biochemical profile.
|
|
| In vivo |
In the preclinical xenograft A2780 human ovarian carcinoma model, this compound reveals good efficacy and is well tolerated upon repeated daily treatments. Treatment of K-Ras(G12D)LA2 mice with this chemical (40 mg/kg twice daily for 10 days) results in significant tumor growth inhibition at the end of the treatment and is accompanied by a reduction in the cell membrane turnover. On the other hand, following oral administration, it shows significant antitumor activity in various human xenografts, carcinogen-induced tumors and in disseminated primary leukemia models; the plasma concentrations in rodents being in the same range as those found inhibiting cancer cell proliferation.
|
References |
|
Applications
| Methods | Biomarkers | Images | PMID |
|---|---|---|---|
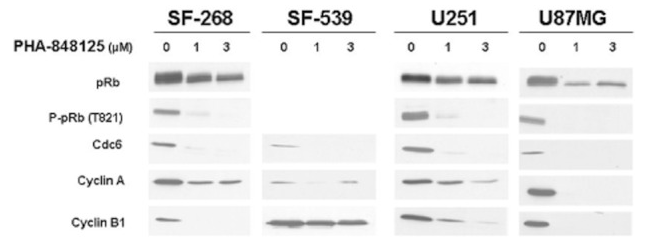
| Western blot | pRb / P-pRb / Cdc6 / Cyclin A / Cyclin B1 p-TrkA / TrkA / p-MAPK / MAPK / p-AKT p-IGF-1Rβ / p-EGFR / p-FGFR3 / p-c-Yes / p-Src / p-S6 |
|
23347136 |
Tech Support
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
Signaling Pathway Map
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.