
- Bioactive Compounds
- By Signaling Pathways
- PI3K/Akt/mTOR
- Epigenetics
- Methylation
- Immunology & Inflammation
- Protein Tyrosine Kinase
- Angiogenesis
- Apoptosis
- Autophagy
- ER stress & UPR
- JAK/STAT
- MAPK
- Cytoskeletal Signaling
- Cell Cycle
- TGF-beta/Smad
- Compound Libraries
- Antibodies
- Bioreagents
- qPCR
- 2x SYBR Green qPCR Master Mix
- 2x SYBR Green qPCR Master Mix(Low ROX)
- 2x SYBR Green qPCR Master Mix(High ROX)
- Protein Assay
- Protein A/G Magnetic Beads for IP
- Anti-Flag magnetic beads
- Anti-Flag Affinity Gel
- Anti-Myc magnetic beads
- Anti-HA magnetic beads
- Poly FLAG Peptide lyophilized powder
- Protease Inhibitor Cocktail
- Protease Inhibitor Cocktail (EDTA-Free, 100X in DMSO)
- Phosphatase Inhibitor Cocktail (2 Tubes, 100X)
- Cell Biology
- Cell Counting Kit-8 (CCK-8)
- Animal Experiment
- Mouse Direct PCR Kit (For Genotyping)
- New Products
- Contact Us
-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
-
 Other Countries
Other Countries
AZD5438
AZD5438 is a potent inhibitor of CDK1/2/9 with IC50 of 16 nM/6 nM/20 nM in cell-free assays. It is less potent to CDK5/6 and also inhibits GSK3β.
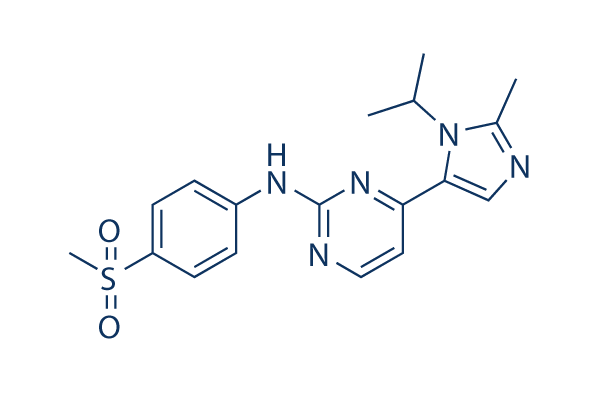
AZD5438 Chemical Structure
CAS: 602306-29-6
Selleck's AZD5438 has been cited by 22 Publications
6 Customer Reviews
Purity & Quality Control
Batch:
Purity:
99.99%
99.99
AZD5438 Related Products
| Related Targets | CDK1 CDK2 CDK3 CDK4 CDK5 CDK6 CDK7 CDK9 CLK Cdc CDK8 CDK12 CDK13 CDK19 CDK11 CDK/cyclin complexes | Click to Expand |
|---|---|---|
| Related Compound Libraries | Kinase Inhibitor Library PI3K/Akt Inhibitor Library MAPK Inhibitor Library DNA Damage/DNA Repair compound Library Cell Cycle compound library | Click to Expand |
Signaling Pathway
Choose Selective CDK Inhibitors
Cell Data
| Cell Lines | Assay Type | Concentration | Incubation Time | Formulation | Activity Description | PMID |
|---|---|---|---|---|---|---|
| human LoVo cells | Proliferation assay | 48 h | Antiproliferative activity against human LoVo cells assessed as BrdU incorporation after 48 hrs, IC50=0.63 μM | 18815031 | ||
| human LoVo cells | Proliferation assay | 48 h | Antiproliferative activity against human LoVo cells assessed as inhibition of BrdU incorporation after 48 hrs, IC50=0.8 μM | 18996007 | ||
| Click to View More Cell Line Experimental Data | ||||||
Biological Activity
| Description | AZD5438 is a potent inhibitor of CDK1/2/9 with IC50 of 16 nM/6 nM/20 nM in cell-free assays. It is less potent to CDK5/6 and also inhibits GSK3β. | ||||||
|---|---|---|---|---|---|---|---|
| Features | A potent inhibitor of cyclin-dependent kinase (CDK) 1, 2, and 9. | ||||||
| Targets |
|
| In vitro | ||||
| In vitro | AZD5438 exhibits the potent inhibitory effect on activity of cyclin-dependent kinases including cyclin E-cdk2, cyclin A-cdk2, cyclin B1-cdk1, cyclin D3-cdk6, and cyclin T-cdk9 with IC50 of 6 nM, 45 nM, 16 nM, 21 nM, and 20 nM, respectively. Besides, AZD5438 also inhibits the kinase activity of p25-cdk5 and glycogen synthase kinase 3β with IC50 of 14 nM and 17 nM, respectively. [1] AZD5438 induces cell cycle arrest by inhibiting phosphorylation of cdk-dependent substrates, and exhibits the broad antiproliferative activity against a range of tumor cell lines including lung, colorectal, breast, prostate, and hematologic tumors with IC50 ranging from 0.2 μM (MCF-7) to 1.7 μM (ARH-77). [1] |
|||
|---|---|---|---|---|
| Kinase Assay | Recombinant Kinase Assays [1] | |||
| The ability of AZD5438 to inhibit cdk activity is examined using a scintillation proximity assay with recombinant cdk-cyclin complexes of cyclin-Ecdk2, cdk2-cyclin A, cdk4-cyclin D, and recombinant retinoblastoma substrate (amino acids 792-928) or cdk1-cyclin B1 with a peptide substrate derived from the in vitro p34cdc2 phosphorylation site of histone H1 (biotin-X-Pro-Lys-Thr-Pro-Lys-Lys-Ala-Lys-Lys-Leu). The activity of AZD5438 against recombinant cdk5/p25 (at 2 μM ATP) is determined in a scintillation proximity assay-based assay using peptide substrate (AKKPKTPKKAKKLOH). Inhibition of glycogen synthase kinase 3β activity is determined with scintillation proximity assay based on the use of human purified glycogen synthase kinase 3βenzyme and eukaryotic initiation factor 2B substrate (at 1 μM ATP). AZD5438 is screened against active recombinant human cdk6-cyclin D3, cdk7-cyclin H/MAT1 (cdk activating kinase complex), and cdk9-cyclin T using the kinase selectivity screening service. | ||||
| Cell Research | Cell lines | MCF-7, HCT-116, A549, and IM-9 | ||
| Concentrations | 0 to 10 μM | |||
| Incubation Time | 48 hours or 72 hours | |||
| Method | AZD5438 is tested against solid tumor cell lines. Briefly, cells are incubated for 48 hours with AZD5438 at a range of concentrations. At the end of incubation, the cells are pulsed with 5-bromo-2′-deoxyuridine (BrdUrd) and the amount of DNA synthesis is measured. The IC50 for inhibition of proliferation is specifically determined independently of cell death. Multiple myeloma cell lines are seeded into 96-well plates in RPMI 1640 supplemented with 10% FCS and glutamine and dosed with AZD5438 for 72 hours. Cell growth is measured using AlamarBlue and GI50 values are calculated with reference to pretreatment control values. |
|||
| Experimental Result Images | Methods | Biomarkers | Images | PMID |
| Western blot | p-Chk2 / Chk2 / p-Chk1 / Chk1 |
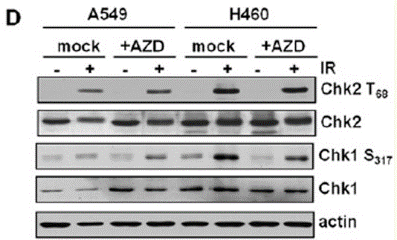
|
22795803 | |
| Growth inhibition assay | Cell viability |
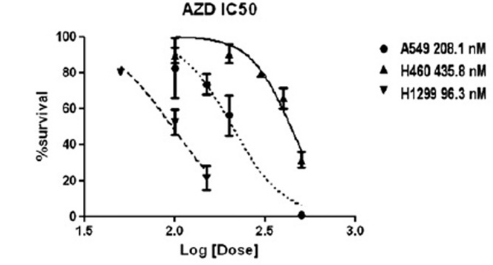
|
22795803 | |
| In Vivo | ||
| In vivo |
In vivo, oral treatment of AZD5438 leads to statistically significant inhibition against the growth of human tumor xenografts derived from a wide range of different cancer types including breast, colon, lung, prostate, and ovarian with maximum TGI ranging from 38% to 153%. [1] In the SW620 xenograft model, AZD5438 causes the inhibition of several cell cycle proteins such as, phH3, phosphonucleolin, PP1a, and several phospho-pRb epitopes in a dose-dependent manner. [1] |
|
|---|---|---|
| Animal Research | Animal Models | MCF-7, HCT-116, A549, and IM-9 cells are injected s.c. into the Swiss nude mice and nude rats. |
| Dosages | ≤100 mg/kg | |
| Administration | Administered via p.o. | |
Chemical Information & Solubility
| Molecular Weight | 371.46 | Formula | C18H21N5O2S |
| CAS No. | 602306-29-6 | SDF | Download AZD5438 SDF |
| Smiles | CC1=NC=C(N1C(C)C)C2=NC(=NC=C2)NC3=CC=C(C=C3)S(=O)(=O)C | ||
| Storage (From the date of receipt) | |||
|
In vitro |
DMSO : 74 mg/mL ( (199.21 mM); Moisture-absorbing DMSO reduces solubility. Please use fresh DMSO.) Ethanol : 74 mg/mL Water : Insoluble |
Molecular Weight Calculator |
|
In vivo Add solvents to the product individually and in order. |
In vivo Formulation Calculator |
||||
Preparing Stock Solutions
Molarity Calculator
In vivo Formulation Calculator (Clear solution)
Step 1: Enter information below (Recommended: An additional animal making an allowance for loss during the experiment)
mg/kg
g
μL
Step 2: Enter the in vivo formulation (This is only the calculator, not formulation. Please contact us first if there is no in vivo formulation at the solubility Section.)
% DMSO
%
% Tween 80
% ddH2O
%DMSO
%
Calculation results:
Working concentration: mg/ml;
Method for preparing DMSO master liquid: mg drug pre-dissolved in μL DMSO ( Master liquid concentration mg/mL, Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug. )
Method for preparing in vivo formulation: Take μL DMSO master liquid, next addμL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O, mix and clarify.
Method for preparing in vivo formulation: Take μL DMSO master liquid, next add μL Corn oil, mix and clarify.
Note: 1. Please make sure the liquid is clear before adding the next solvent.
2. Be sure to add the solvent(s) in order. You must ensure that the solution obtained, in the previous addition, is a clear solution before proceeding to add the next solvent. Physical methods such
as vortex, ultrasound or hot water bath can be used to aid dissolving.
Tech Support
Answers to questions you may have can be found in the inhibitor handling instructions. Topics include how to prepare stock solutions, how to store inhibitors, and issues that need special attention for cell-based assays and animal experiments.
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
* Indicates a Required Field
Tags: buy AZD5438 | AZD5438 supplier | purchase AZD5438 | AZD5438 cost | AZD5438 manufacturer | order AZD5438 | AZD5438 distributor
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.