-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
research use only
Foretinib (GSK1363089, XL880) Multi-targeted Tyrosine Kinase Inhibitor
Cat.No.S1111
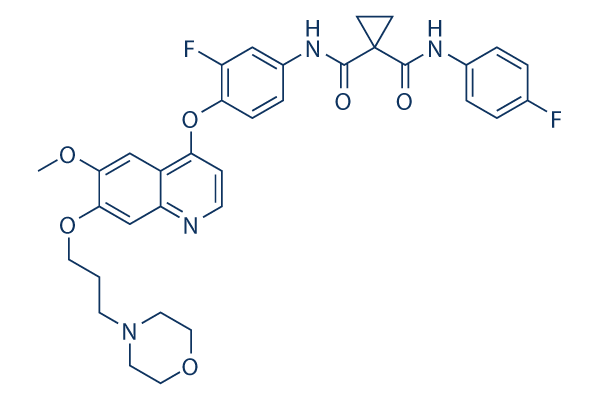
Chemical Structure
Molecular Weight: 632.65
Quality Control
| Related Targets | EGFR VEGFR PDGFR FGFR Src MEK CSF-1R FLT3 HER2 c-Kit |
|---|---|
| Other c-Met Inhibitors | Tepotinib (EMD-1214063) Dihexa SGX-523 PHA-665752 SU11274 BMS-777607 JNJ-38877605 Tivantinib PF-04217903 Savolitinib (AZD6094) |
Cell Culture, Treatment & Working Concentration
| Cell Lines | Assay Type | Concentration | Incubation Time | Formulation | Activity Description | PMID |
|---|---|---|---|---|---|---|
| Daoy | Function Assay | 0.5/1/2.5 μM | 24 h | DMSO | inhibits the HGF-induced cMET pathway activation | 25391241 |
| ONS76 | Function Assay | 0.5/1/2.5 μM | 24 h | DMSO | inhibits the HGF-induced cMET pathway activation | 25391241 |
| Daoy | Function Assay | 0.5/1/2.5 μM | 24 h | DMSO | inhibits HGF-mediated migration and invasion | 25391241 |
| ONS76 | Function Assay | 0.5/1/2.5 μM | 24 h | DMSO | inhibits HGF-mediated migration and invasion | 25391241 |
| Daoy | Apoptosis Assay | 1 μM | 24 h | DMSO | induces apoptosis | 25391241 |
| Daoy | Growth Inhibition Assay | 0.5/1/2.5 μM | 24-96 h | DMSO | inhibits cell growth in a dose dependent manner | 25391241 |
| U251 | Function Assay | 100/300/900 nM | 1 h | DMSO | inhibits the phosphorylation of MerTK | 24658326 |
| A172 | Function Assay | 100/300/900 nM | 1 h | DMSO | inhibits the phosphorylation of MerTK | 24658326 |
| SF188 | Function Assay | 100/300/900 nM | 1 h | DMSO | inhibits the phosphorylation of MerTK | 24658326 |
| U251 | Function Assay | 100/300/900 nM | 1 h | DMSO | inhibits the activity of Axl, Tyro3 | 24658326 |
| A172 | Function Assay | 100/300/900 nM | 1 h | DMSO | inhibits the activity of Axl, Tyro3 | 24658326 |
| SF188 | Function Assay | 100/300/900 nM | 1 h | DMSO | inhibits the activity of Axl, Tyro3 | 24658326 |
| U251 | Function Assay | 100/300/900 nM | 1 h | DMSO | decreases Akt phosphorylation in a concentration dependent manner | 24658326 |
| A172 | Function Assay | 100/300/900 nM | 1 h | DMSO | decreases Akt phosphorylation in a concentration dependent manner | 24658326 |
| SF188 | Function Assay | 100/300/900 nM | 1 h | DMSO | decreases Akt phosphorylation in a concentration dependent manner | 24658326 |
| U251 | Function Assay | 100/300/900 nM | 28 h | DMSO | induces PARP cleavage | 24658326 |
| SF188 | Function Assay | 100/300/900 nM | 28 h | DMSO | induces PARP cleavage | 24658326 |
| SF188 | Growth Inhibition Assay | 100/300/900 nM | 48 h | DMSO | reduces cell survival at 900 nM significantly | 24658326 |
| U251 | Growth Inhibition Assay | 100/300/900 nM | 48 h | DMSO | reduces cell survival at 900 nM significantly | 24658326 |
| A172 | Growth Inhibition Assay | 100/300/900 nM | 48 h | DMSO | reduces cell survival at 900 nM significantly | 24658326 |
| SF188 | Function Assay | 100/300/900 nM | 24 h | DMSO | abrogates migration and invasion of glioma cells in a dose dependent manner | 24658326 |
| U251 | Function Assay | 100/300/900 nM | 24 h | DMSO | abrogates migration and invasion of glioma cells in a dose dependent manner | 24658326 |
| A172 | Function Assay | 100/300/900 nM | 24 h | DMSO | abrogates migration and invasion of glioma cells in a dose dependent manner | 24658326 |
| Ba/F3 | Cell Viability Assay | 0.0001-10 μM | 72 h | inhibits cell growth in a dose dependent manner | 24218589 | |
| HCC78 | Cell Viability Assay | 0.01-10 μM | 72 h | inhibits cell growth in a dose dependent manner | 24218589 | |
| SK-HEP1 | Cell Viability Assay | 0.25-1.5 μM | 24 h | inhibits cell growth in a dose dependent manner | 22187171 | |
| SK-HEP2 | Function Assay | 1 μM | 24 h | blocks HGF-induced cell motility | 22187171 | |
| SK-HEP2 | Function Assay | 1 μM | 24 h | causes G2/M phase arrest with reduction in the G0/G1 and S phases | 22187171 | |
| MKN-45 | Growth Inhibition Assay | 0.01-10 μM | 5 d | IC50=8 nM | 21655918 | |
| KATO-III | Growth Inhibition Assay | 0.01-10 μM | 5 d | IC50=30 nM | 21655918 | |
| MKN-45 | Function Assay | 1 μM | 24 h | inhibits phosphorylation of MET, Akt, and ERK1/2 in MKN-45 | 21655918 | |
| KATO-III | Function Assay | 1 μM | 24 h | inhibits phosphorylation of MET, Akt, and ERK1/2 in MKN-45 | 21655918 | |
| H1648 | Growth Inhibition Assay | IC50=1.28 ±0.12 μM | 21252284 | |||
| H1573 | Growth Inhibition Assay | IC50=1.62 ± 0.05 μM | 21252284 | |||
| H596 | Growth Inhibition Assay | IC50=1.21 ± 0.17 μM | 21252284 | |||
| HOP92 | Growth Inhibition Assay | IC50=0.81 ± 0.29 μM | 21252284 | |||
| H69 | Growth Inhibition Assay | IC50=1.18 ± 0.08 μM | 21252284 | |||
| H1975 | Growth Inhibition Assay | IC50=1.39 ± 0.33 μM | 21252284 | |||
| SCC15 | Growth Inhibition Assay | IC50=0.63 ± 0.04 μM | 21252284 | |||
| HN5 | Growth Inhibition Assay | IC50=0.65 ± 0.26 μM | 21252284 | |||
| MKN45 | Cytotoxicity assay | 72 hrs | Cytotoxicity against c-Met-dependent human MKN45 cells assessed as growth inhibition after 72 hrs by MTT assay, IC50 = 0.03 μM. | 23628470 | ||
| A549 | Cytotoxicity assay | 72 hrs | Cytotoxicity against c-Met-dependent human A549 cells assessed as growth inhibition after 72 hrs by MTT assay, IC50 = 0.08 μM. | 23628470 | ||
| HT-29 | Cytotoxicity assay | 72 hrs | Cytotoxicity against c-Met-dependent human HT-29 cells assessed as growth inhibition after 72 hrs by MTT assay, IC50 = 0.15 μM. | 23628470 | ||
| H460 | Cytotoxicity assay | 72 hrs | Cytotoxicity against c-Met-independent human H460 cells assessed as growth inhibition after 72 hrs by MTT assay, IC50 = 0.18 μM. | 23628470 | ||
| SMMC7721 | Cytotoxicity assay | 72 hrs | Cytotoxicity against c-Met-dependent human SMMC7721 cells assessed as growth inhibition after 72 hrs by MTT assay, IC50 = 0.44 μM. | 23628470 | ||
| U87MG | Cytotoxicity assay | 72 hrs | Cytotoxicity against c-Met-dependent human U87MG cells assessed as growth inhibition after 72 hrs by MTT assay, IC50 = 0.9 μM. | 23628470 | ||
| MKN45 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MKN45 cells after 72 hrs by MTT assay, IC50 = 0.031 μM. | 23644189 | ||
| H460 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human H460 cells after 72 hrs by MTT assay, IC50 = 0.12 μM. | 23644189 | ||
| HT-29 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HT-29 cells after 72 hrs by MTT assay, IC50 = 0.18 μM. | 23644189 | ||
| SMMC7721 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human SMMC7721 cells after 72 hrs by MTT assay, IC50 = 0.3 μM. | 23644189 | ||
| U87MG | Cytotoxicity assay | 72 hrs | Cytotoxicity against human U87MG cells after 72 hrs by MTT assay, IC50 = 1.04 μM. | 23644189 | ||
| MKN45 | Cytotoxicity assay | Cytotoxicity against human MKN45 cells, IC50 = 0.008 μM. | 23838381 | |||
| MKN45 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MKN45 cells after 72 hrs by MTT assay, IC50 = 0.023 μM. | 23838381 | ||
| A549 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human A549 cells after 72 hrs by DAPI staining-based fluorescence assay, IC50 = 0.029 μM. | 23838381 | ||
| HT-29 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HT-29 cells after 72 hrs by DAPI staining-based fluorescence assay, IC50 = 0.165 μM. | 23838381 | ||
| A549 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human A549 cells after 72 hrs by MTT assay, IC50 = 0.17 μM. | 23838381 | ||
| NCI-H460 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human NCI-H460 cells after 72 hrs by MTT assay, IC50 = 0.21 μM. | 23838381 | ||
| HT-29 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HT-29 cells after 72 hrs by MTT assay, IC50 = 0.26 μM. | 23838381 | ||
| SMMC7721 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human SMMC7721 cells after 72 hrs by MTT assay, IC50 = 0.32 μM. | 23838381 | ||
| U87MG | Cytotoxicity assay | 72 hrs | Cytotoxicity against human U87MG cells after 72 hrs by MTT assay, IC50 = 0.47 μM. | 23838381 | ||
| MKN45 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MKN45 cells after 72 hrs by MTT assay, IC50 = 0.032 μM. | 24012712 | ||
| A549 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human A549 cells after 72 hrs by MTT assay, IC50 = 0.11 μM. | 24012712 | ||
| HT-29 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HT-29 cells after 72 hrs by MTT assay, IC50 = 0.19 μM. | 24012712 | ||
| H460 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human H460 cells after 72 hrs by MTT assay, IC50 = 0.21 μM. | 24012712 | ||
| U87MG | Cytotoxicity assay | 72 hrs | Cytotoxicity against human U87MG cells after 72 hrs by MTT assay, IC50 = 1.08 μM. | 24012712 | ||
| MKN45 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MKN45 cells after 72 hrs by MTT assay, IC50 = 0.032 μM. | 24485123 | ||
| A549 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human A549 cells after 72 hrs by MTT assay, IC50 = 0.13 μM. | 24485123 | ||
| HT-29 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HT-29 cells after 72 hrs by MTT assay, IC50 = 0.16 μM. | 24485123 | ||
| H460 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human H460 cells after 72 hrs by MTT assay, IC50 = 0.19 μM. | 24485123 | ||
| U87MG | Cytotoxicity assay | 72 hrs | Cytotoxicity against human U87MG cells after 72 hrs by MTT assay, IC50 = 1.1 μM. | 24485123 | ||
| MKN45 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MKN45 cells after 72 hrs by MTT assay, IC50 = 0.032 μM. | 24882675 | ||
| HT-29 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HT-29 cells after 72 hrs by MTT assay, IC50 = 0.19 μM. | 24882675 | ||
| H460 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human H460 cells after 72 hrs by MTT assay, IC50 = 0.21 μM. | 24882675 | ||
| MDA-MB-231 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MDA-MB-231 cells after 72 hrs by MTT assay, IC50 = 0.54 μM. | 24882675 | ||
| MKN45 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MKN45 cells assessed as inhibition of cell growth after 72 hrs by MTT assay, IC50 = 0.032 μM. | 24996144 | ||
| A549 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human A549 cells assessed as inhibition of cell growth after 72 hrs by MTT assay, IC50 = 0.11 μM. | 24996144 | ||
| HT-29 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HT-29 cells assessed as inhibition of cell growth after 72 hrs by MTT assay, IC50 = 0.19 μM. | 24996144 | ||
| H460 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human H460 cells assessed as inhibition of cell growth after 72 hrs by MTT assay, IC50 = 0.21 μM. | 24996144 | ||
| U87MG | Cytotoxicity assay | 72 hrs | Cytotoxicity against human U87MG cells assessed as inhibition of cell growth after 72 hrs by MTT assay, IC50 = 1.08 μM. | 24996144 | ||
| MKN45 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human MKN45 cells assessed as inhibition of cell growth after 72 hrs by MTT assay, IC50 = 0.032 μM. | 25282672 | ||
| A549 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human A549 cells assessed as inhibition of cell growth after 72 hrs by MTT assay, IC50 = 0.12 μM. | 25282672 | ||
| HT-29 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human HT-29 cells assessed as inhibition of cell growth after 72 hrs by MTT assay, IC50 = 0.19 μM. | 25282672 | ||
| H460 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human H460 cells assessed as inhibition of cell growth after 72 hrs by MTT assay, IC50 = 0.21 μM. | 25282672 | ||
| U87MG | Antiproliferative assay | 72 hrs | Antiproliferative activity against human U87MG cells assessed as inhibition of cell growth after 72 hrs by MTT assay, IC50 = 1.02 μM. | 25282672 | ||
| MKN45 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MKN45 cells after 72 hrs by MTT assay, IC50 = 0.032 μM. | 25438768 | ||
| A549 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human A549 cells after 72 hrs by MTT assay, IC50 = 0.13 μM. | 25438768 | ||
| HT-29 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HT-29 cells after 72 hrs by MTT assay, IC50 = 0.16 μM. | 25438768 | ||
| H460 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human H460 cells after 72 hrs by MTT assay, IC50 = 0.19 μM. | 25438768 | ||
| U87MG | Cytotoxicity assay | 72 hrs | Cytotoxicity against human U87MG cells after 72 hrs by MTT assay, IC50 = 0.92 μM. | 25438768 | ||
| BA/F3 | Function assay | Inhibition of ROS1 (unknown origin) expressed in mouse BA/F3 cells, IC50 = 0.01 μM. | 25461320 | |||
| HT-29 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HT-29 cells assessed as inhibition of cell proliferation after 72 hrs by MTT assay, IC50 = 0.032 μM. | 26169763 | ||
| MKN45 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MKN45 cells assessed as inhibition of cell proliferation after 72 hrs by MTT assay, IC50 = 0.12 μM. | 26169763 | ||
| A549 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human A549 cells assessed as inhibition of cell proliferation after 72 hrs by MTT assay, IC50 = 0.19 μM. | 26169763 | ||
| H460 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human H460 cells assessed as inhibition of cell proliferation after 72 hrs by MTT assay, IC50 = 0.21 μM. | 26169763 | ||
| BT474 | Function assay | Inhibition of AXL in lapatinib-sensitive human BT474 cells, IC50 = 0.1 μM. | 26555154 | |||
| PC3 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human PC3 cells after 72 hrs by MTT assay, IC50 = 0.39 μM. | 26810712 | ||
| A549 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human A549 cells after 72 hrs by MTT assay, IC50 = 0.64 μM. | 26810712 | ||
| MCF7 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MCF7 cells after 72 hrs by MTT assay, IC50 = 9.47 μM. | 26810712 | ||
| MKN45 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MKN45 cells after 72 hrs by MTT assay, IC50 = 0.023 μM. | 26897090 | ||
| A549 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human A549 cells after 72 hrs by MTT assay, IC50 = 0.17 μM. | 26897090 | ||
| HT-29 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HT-29 cells after 72 hrs by MTT assay, IC50 = 0.26 μM. | 26897090 | ||
| HT-29 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HT-29 cells assessed as reduction in cell growth after 72 hrs by MTT assay, IC50 = 0.26 μM. | 26923692 | ||
| A549 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human A549 cells assessed as reduction in cell growth after 72 hrs by MTT assay, IC50 = 0.49 μM. | 26923692 | ||
| PC3 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human PC3 cells assessed as reduction in cell growth after 72 hrs by MTT assay, IC50 = 0.89 μM. | 26923692 | ||
| MCF7 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MCF7 cells assessed as reduction in cell growth after 72 hrs by MTT assay, IC50 = 6.25 μM. | 26923692 | ||
| MKN45 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MKN45 cells assessed as cell growth inhibition after 72 hrs by MTT assay, IC50 = 0.032 μM. | 26944614 | ||
| A549 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human A549 cells assessed as cell growth inhibition after 72 hrs by MTT assay, IC50 = 0.13 μM. | 26944614 | ||
| HT-29 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HT-29 cells assessed as cell growth inhibition after 72 hrs by MTT assay, IC50 = 0.16 μM. | 26944614 | ||
| H460 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human H460 cells assessed as cell growth inhibition after 72 hrs by MTT assay, IC50 = 0.19 μM. | 26944614 | ||
| U87MG | Cytotoxicity assay | 72 hrs | Cytotoxicity against human U87MG cells assessed as cell growth inhibition after 72 hrs by MTT assay, IC50 = 1.1 μM. | 26944614 | ||
| PC3 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human PC3 cells after 72 hrs by MTT assay, IC50 = 0.39 μM. | 26964675 | ||
| A549 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human A549 cells after 72 hrs by MTT assay, IC50 = 0.64 μM. | 26964675 | ||
| MCF7 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MCF7 cells after 72 hrs by MTT assay, IC50 = 9.47 μM. | 26964675 | ||
| BA/F3 | Antiproliferative assay | 72 hrs | Antiproliferative activity against mouse BA/F3 cells expressing TPR-Met after 72 hrs by CCK8 assay, IC50 = 0.0092 μM. | 27068889 | ||
| MKN45 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MKN45 cells measured after 72 hrs by MTT assay, IC50 = 0.032 μM. | 27155466 | ||
| A549 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human A549 cells measured after 72 hrs by MTT assay, IC50 = 0.11 μM. | 27155466 | ||
| HT-29 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HT-29 cells measured after 72 hrs by MTT assay, IC50 = 0.19 μM. | 27155466 | ||
| H460 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human H460 cells measured after 72 hrs by MTT assay, IC50 = 0.21 μM. | 27155466 | ||
| MKN45 | Cytotoxicity assay | Cytotoxicity against human MKN45 cells, IC50 = 0.008 μM. | 27187857 | |||
| MKN45 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MKN45 cells after 72 hrs by MTT assay, IC50 = 0.023 μM. | 27187857 | ||
| HT-29 | Cytotoxicity assay | Cytotoxicity against human HT-29 cells, IC50 = 0.029 μM. | 27187857 | |||
| A549 | Cytotoxicity assay | Cytotoxicity against human A549 cells, IC50 = 0.165 μM. | 27187857 | |||
| HT-29 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HT-29 cells after 72 hrs by MTT assay, IC50 = 0.17 μM. | 27187857 | ||
| A549 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human A549 cells after 72 hrs by MTT assay, IC50 = 0.26 μM. | 27187857 | ||
| MDA-MB-231 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MDA-MB-231 cells after 72 hrs by MTT assay, IC50 = 0.54 μM. | 27187857 | ||
| Sf9 | Function assay | 60 mins | Inhibition of NH2-terminal His-tagged MET kinase domain (1051 to 1348 residues) (unknown origin) expressed in baculovirus infected Sf9 insect cells preincubated for 60 mins followed by (poly(Glu, Tyr) 4:1) substrate addition for 2 to 4 hrs by luciferase-c, IC50 = 0.0004 μM. | 27299736 | ||
| MKN45 | Cytotoxicity assay | 72 hrs | Cytotoxicity against c-Met over-expressed human MKN45 cells assessed as cell growth inhibition after 72 hrs by MTT assay, IC50 = 0.032 μM. | 27490023 | ||
| A549 | Cytotoxicity assay | 72 hrs | Cytotoxicity against c-Met over-expressed human A549 cells assessed as cell growth inhibition after 72 hrs by MTT assay, IC50 = 0.14 μM. | 27490023 | ||
| HT-29 | Cytotoxicity assay | 72 hrs | Cytotoxicity against c-Met over-expressed human HT-29 cells assessed as cell growth inhibition after 72 hrs by MTT assay, IC50 = 0.19 μM. | 27490023 | ||
| H460 | Cytotoxicity assay | 72 hrs | Cytotoxicity against c-Met over-expressed human H460 cells assessed as cell growth inhibition after 72 hrs by MTT assay, IC50 = 0.21 μM. | 27490023 | ||
| A549 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human A549 cells after 72 hrs by MTT assay, IC50 = 0.15 μM. | 28011202 | ||
| HT-29 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HT-29 cells after 72 hrs by MTT assay, IC50 = 0.25 μM. | 28011202 | ||
| MKN45 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MKN45 cells after 72 hrs by MTT assay, IC50 = 0.32 μM. | 28011202 | ||
| HT-29 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HT-29 cells after 72 hrs by MTT assay, IC50 = 0.25 μM. | 28384549 | ||
| H460 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human H460 cells after 72 hrs by MTT assay, IC50 = 0.29 μM. | 28384549 | ||
| A549 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human A549 cells after 72 hrs by MTT assay, IC50 = 0.36 μM. | 28384549 | ||
| U87MG | Cytotoxicity assay | 72 hrs | Cytotoxicity against human U87MG cells after 72 hrs by MTT assay, IC50 = 0.96 μM. | 28384549 | ||
| MKN45 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human MKN45 cells after 72 hrs by MTT assay, IC50 = 0.03 μM. | 28716639 | ||
| A549 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human A549 cells after 72 hrs by MTT assay, IC50 = 0.15 μM. | 28716639 | ||
| HT-29 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human HT-29 cells after 72 hrs by MTT assay, IC50 = 0.21 μM. | 28716639 | ||
| H460 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human H460 cells after 72 hrs by MTT assay, IC50 = 0.22 μM. | 28716639 | ||
| U87MG | Antiproliferative assay | 72 hrs | Antiproliferative activity against human U87MG cells after 72 hrs by MTT assay, IC50 = 0.96 μM. | 28716639 | ||
| HCC827 | Function assay | Displacement of [3H]-cyclopamine from SMO V404M mutant in gefitinib resistant human HCC827 cells by scintillation counting, Ki = 0.0668 μM. | 28787156 | |||
| HCC827 | Antiproliferative assay | 72 hrs | Antiproliferative activity against gefitinib resistant EGFR-mutated human HCC827 cells after 72 hrs by MTT assay, IC50 = 0.5 μM. | 28787156 | ||
| HCC827 | Antitumor assay | 15 mg/kg | Antitumor activity against gefitinib resistant human HCC827 cells xenografted in balb/c athymic nu/nu mouse assessed as decrease in tumor size at 15 mg/kg/day, po | 28787156 | ||
| HCC827 | Apoptosis assay | 0.01 to 1 uM | 72 hrs | Induction of apoptosis in gefitinib resistant human HCC827 cells at 0.01 to 1 uM after 72 hrs by annexin V-FITC-propidium iodide staining-based flow cytometry method relative to control | 28787156 | |
| HCC827 | Function assay | 0.1 to 0.5 uM | 72 hrs | Inhibition of MET phosphorylation at Tyr1234/1235 in gefitinib resistant human HCC827 cells at 0.1 to 0.5 uM after 72 hrs by Western blotting method | 28787156 | |
| HCC827 | Function assay | 0.1 to 0.5 uM | 72 hrs | Downregulation of SMO expression in gefitinib resistant human HCC827 cells at 0.1 to 0.5 uM after 72 hrs by Western blotting method | 28787156 | |
| HCC827 | Function assay | 0.1 to 0.5 uM | 72 hrs | Inhibition of MET in gefitinib resistant human HCC827 cells assessed as decrease in MAPK44/42 phosphorylation at Thr202/Tyr204 at 0.1 to 0.5 uM after 72 hrs by Western blotting method | 28787156 | |
| HCC827 | Function assay | 0.1 to 0.5 uM | 72 hrs | Inhibition of MET in gefitinib resistant human HCC827 cells assessed as decrease in AKT phosphorylation at Ser473 at 0.1 to 0.5 uM after 72 hrs by Western blotting method | 28787156 | |
| HCC827 | Function assay | 0.1 to 0.5 uM | 72 hrs | Inhibition of MET/SMO V404M mutant in gefitinib resistant human HCC827 cells assessed as decrease in vimentin expression at 0.1 to 0.5 uM after 72 hrs by Western blotting method | 28787156 | |
| HCC827 | Antitumor assay | 20 mg/kg | Antitumor activity against gefitinib resistant human HCC827 cells xenografted in balb/c athymic nu/nu mouse assessed as decrease in tumor size at 20 mg/kg/day, po | 28787156 | ||
| HCC827 | Antitumor assay | 20 mg/kg | Antitumor activity against gefitinib resistant human HCC827 cells xenografted in balb/c athymic nu/nu mouse assessed as decrease in tumor size at 20 mg/kg/day, po in presence of gefitinib | 28787156 | ||
| HCC827 | Antitumor assay | 20 mg/kg | Antitumor activity against gefitinib resistant human HCC827 cells xenografted in balb/c athymic nu/nu mouse assessed as decrease in tumor size at 20 mg/kg/day, po in presence of osimertinib | 28787156 | ||
| PC3 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human PC3 cells assessed as reduction in cell viability after 72 hrs by MTT assay, IC50 = 1.48 μM. | 29107421 | ||
| A549 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human A549 cells assessed as reduction in cell viability after 72 hrs by MTT assay, IC50 = 1.74 μM. | 29107421 | ||
| HepG2 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HepG2 cells assessed as reduction in cell viability after 72 hrs by MTT assay, IC50 = 2.88 μM. | 29107421 | ||
| MCF7 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MCF7 cells assessed as reduction in cell viability after 72 hrs by MTT assay, IC50 = 3.92 μM. | 29107421 | ||
| HT-29 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human HT-29 cells after 72 hrs by MTT assay, IC50 = 0.26 μM. | 29197731 | ||
| NCI-H460 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human NCI-H460 cells after 72 hrs by MTT assay, IC50 = 0.28 μM. | 29197731 | ||
| A549 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human A549 cells after 72 hrs by MTT assay, IC50 = 0.32 μM. | 29197731 | ||
| U87MG | Antiproliferative assay | 72 hrs | Antiproliferative activity against human U87MG cells after 72 hrs by MTT assay, IC50 = 0.91 μM. | 29197731 | ||
| HepG2 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HepG2 cells after 72 hrs by MTT assay, IC50 = 2.27 μM. | 29203143 | ||
| MCF7 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MCF7 cells after 72 hrs by MTT assay, IC50 = 2.76 μM. | 29203143 | ||
| A549 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human A549 cells after 72 hrs by MTT assay, IC50 = 3.13 μM. | 29203143 | ||
| PC3 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human PC3 cells after 72 hrs by MTT assay, IC50 = 0.39 μM. | 29331754 | ||
| A549 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human A549 cells after 72 hrs by MTT assay, IC50 = 0.64 μM. | 29331754 | ||
| MCF7 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human MCF7 cells after 72 hrs by MTT assay, IC50 = 9.47 μM. | 29331754 | ||
| TC32 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for TC32 cells | 29435139 | |||
| DAOY | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for DAOY cells | 29435139 | |||
| SJ-GBM2 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SJ-GBM2 cells | 29435139 | |||
| A673 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for A673 cells | 29435139 | |||
| SK-N-MC | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SK-N-MC cells | 29435139 | |||
| BT-37 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for BT-37 cells | 29435139 | |||
| NB-EBc1 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for NB-EBc1 cells | 29435139 | |||
| U-2 OS | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for U-2 OS cells | 29435139 | |||
| Saos-2 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for Saos-2 cells | 29435139 | |||
| SK-N-SH | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SK-N-SH cells | 29435139 | |||
| NB1643 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for NB1643 cells | 29435139 | |||
| LAN-5 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for LAN-5 cells | 29435139 | |||
| BT-12 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for BT-12 cells | 29435139 | |||
| Rh18 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for Rh18 cells | 29435139 | |||
| OHS-50 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for OHS-50 cells | 29435139 | |||
| RD | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for RD cells | 29435139 | |||
| MG 63 (6-TG R) | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for MG 63 (6-TG R) cells | 29435139 | |||
| fibroblast cells | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for control Hh wild type fibroblast cells | 29435139 | |||
| Rh30 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for Rh30 cells | 29435139 | |||
| Rh41 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for Rh41 cells | 29435139 | |||
| NB1643 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for NB1643 cells | 29435139 | |||
| A673 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for A673 cells) | 29435139 | |||
| SK-N-MC | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for SK-N-MC cells | 29435139 | |||
| BT-12 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for BT-12 cells | 29435139 | |||
| LAN-5 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for LAN-5 cells | 29435139 | |||
| DAOY | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for DAOY cells | 29435139 | |||
| NB-EBc1 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for NB-EBc1 cells | 29435139 | |||
| SJ-GBM2 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for SJ-GBM2 cells | 29435139 | |||
| BT-37 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for BT-37 cells | 29435139 | |||
| TC32 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for TC32 cells | 29435139 | |||
| MG 63 (6-TG R) | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for MG 63 (6-TG R) cells | 29435139 | |||
| fibroblast cells | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for control Hh wild type fibroblast cells | 29435139 | |||
| U-2 OS | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for U-2 OS cells | 29435139 | |||
| Rh41 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for Rh41 cells | 29435139 | |||
| RD | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for RD cells | 29435139 | |||
| Rh18 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for Rh18 cells | 29435139 | |||
| Saos-2 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for Saos-2 cells | 29435139 | |||
| OHS-50 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for OHS-50 cells | 29435139 | |||
| SK-N-SH | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Confirmatory screen for SK-N-SH cells | 29435139 | |||
| A549 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human A549 cells after 72 hrs by MTT assay, IC50 = 0.32 μM. | 30216852 | ||
| HepG2 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human HepG2 cells after 72 hrs by MTT assay, IC50 = 0.48 μM. | 30216852 | ||
| MCF7 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human MCF7 cells after 72 hrs by MTT assay, IC50 = 0.76 μM. | 30216852 | ||
| MKN45 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human MKN45 cells after 72 hrs by MTT assay, IC50 = 0.0023 μM. | 30248654 | ||
| EBC1 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human EBC1 cells after 72 hrs by MTT assay, IC50 = 0.0048 μM. | 30248654 | ||
| SNU5 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human SNU5 cells after 72 hrs by MTT assay, IC50 = 0.0089 μM. | 30248654 | ||
| BAF3 | Function assay | 72 hrs | Inhibition of TPR-tagged met (unknown origin) expressed in mouse BAF3 cells assessed as inhibition of cell proliferation after 72 hrs by MTT assay, IC50 = 0.0092 μM. | 30248654 | ||
| NCI-H460 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human NCI-H460 cells after 72 hrs by MTT assay, IC50 = 0.0396 μM. | 30248654 | ||
| MKN45 | Cytotoxicity assay | 72 hrs | Cytotoxicity in human MKN45 cells assessed as reduction in cell viability incubated for 72 hrs by MTT assay, IC50 = 0.041 μM. | 31079967 | ||
| NCI-H460 | Cytotoxicity assay | 72 hrs | Cytotoxicity in human NCI-H460 cells assessed as reduction in cell viability incubated for 72 hrs by MTT assay, IC50 = 0.18 μM. | 31079967 | ||
| HT-29 | Cytotoxicity assay | 72 hrs | Cytotoxicity in human HT-29 cells assessed as reduction in cell viability incubated for 72 hrs by MTT assay, IC50 = 0.22 μM. | 31079967 | ||
| MDA-MB-231 | Cytotoxicity assay | 72 hrs | Cytotoxicity in human MDA-MB-231 cells assessed as reduction in cell viability incubated for 72 hrs by MTT assay, IC50 = 0.52 μM. | 31079967 | ||
| sf21 | Function assay | 60 mins | Inhibition of wild type N-terminal NH-tagged and avi-tagged dephosphorylated c-MET (956 to 1390 residues) (unknown origin) expressed in sf21 cells using poly (Glu,Tyr) as substrate measured after 60 mins by ADP-Glo kinase assay, IC50 = 0.002512 μM. | 31531204 | ||
| sf21 | Function assay | 60 mins | Inhibition of N-terminal NH-tagged and avi-tagged dephosphorylated c-MET D1228V mutant (956 to 1390 residues) (unknown origin) expressed in sf21 cells using poly (Glu,Tyr) as substrate measured after 60 mins by ADP-Glo kinase assay, IC50 = 0.002512 μM. | 31531204 | ||
| NCI-H1993 | Growth inhibition assay | 3 days | Growth inhibition of CRISPR/Cas9 modified human NCI-H1993 cells harboring cMET D1228V mutant incubated for 3 days by cell titer-glo luminescent cell viability assay, GI50 = 0.05248 μM. | 31531204 | ||
| NCI-H1993 | Growth inhibition assay | 3 days | Growth inhibition of wild type human NCI-H1993 cells incubated for 3 days by cell titer-glo luminescent cell viability assay, GI50 = 0.06166 μM. | 31531204 | ||
| NCI-H1993 | Function assay | 4 hrs | Inhibition of c-Met phosphorylation at Tyr 1234/Tyr1235 residues in human NCI-H1993 cells incubated for 4 hrs by HTRF assay, IC50 = 0.2138 μM. | 31531204 | ||
| NCI-H1993 | Function assay | 4 hrs | Inhibition of c-MET D1228V mutant phosphorylation at Tyr1234/Tyr1235 residues in CRISPR/Cas9 modified human NCI-H1993 cells incubated for 4 hrs by HTRF assay, IC50 = 0.2138 μM. | 31531204 | ||
| NCI-H1993 | Function assay | 1 uM | 4 hrs | Inhibition of c-MET D1228V mutant in CRISPR/Cas9 modified human NCI-H1993 cells assessed as reduction in AKT phosphorylation at 1 uM after 4 hrs by Western blot analysis | 31531204 | |
| NCI-H1993 | Function assay | 1 uM | 4 hrs | Inhibition of c-Met phosphorylation in human NCI-H1993 cells at 1 uM after 4 hrs by Western blot analysis | 31531204 | |
| NCI-1993 | Function assay | 1 uM | 4 hrs | Inhibition of c-MET in human NCI-1993 cells assessed as reduction in AKT phosphorylation at 1 uM after 4 hrs by Western blot analysis | 31531204 | |
| NCI-H1993 | Function assay | 1 uM | 4 hrs | Inhibition of c-Met D1228V mutant phosphorylation in CRISPR/Cas9 modified human NCI-H1993 cells at 1 uM after 4 hrs by Western blot analysis | 31531204 | |
| sf21 | Function assay | Binding affinity to wild type N-terminal NH-tagged and avi-tagged dephosphorylated c-MET (956 to 1390 residues) (unknown origin) expressed in sf21 cells by SPR analysis | 31531204 | |||
| sf21 | Function assay | Binding affinity to N-terminal NH-tagged and avi-tagged dephosphorylated c-MET D1228V mutant (956 to 1390 residues) (unknown origin) expressed in sf21 cells by SPR analysis | 31531204 | |||
| A549 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human A549 cells assessed as reduction in cell viability after 72 hrs by MTT assay, IC50 = 2.4 μM. | 31546197 | ||
| MCF7 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human MCF7 cells assessed as reduction in cell viability after 72 hrs by MTT assay, IC50 = 2.4 μM. | 31546197 | ||
| HepG2 | Antiproliferative assay | 72 hrs | Antiproliferative activity against human HepG2 cells assessed as reduction in cell viability after 72 hrs by MTT assay, IC50 = 2.4 μM. | 31546197 | ||
| MKN45 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human MKN45 cells assessed as reduction in cell viability incubated for 72 hrs by MTT assay, IC50 = 0.2 μM. | 31629631 | ||
| H460 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human H460 cells assessed as reduction in cell viability incubated for 72 hrs by MTT assay, IC50 = 1.14 μM. | 31629631 | ||
| A549 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human A549 cells assessed as reduction in cell viability incubated for 72 hrs by MTT assay, IC50 = 1.49 μM. | 31629631 | ||
| HT-29 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human HT-29 cells assessed as reduction in cell viability incubated for 72 hrs by MTT assay, IC50 = 1.76 μM. | 31629631 | ||
| U87MG | Cytotoxicity assay | 72 hrs | Cytotoxicity against human U87MG cells assessed as reduction in cell viability incubated for 72 hrs by MTT assay, IC50 = 2.17 μM. | 31629631 | ||
| Click to View More Cell Line Experimental Data | ||||||
Solubility
|
In vitro |
DMSO
: 100 mg/mL
(158.06 mM)
Water : Insoluble Ethanol : Insoluble |
Molarity Calculator
|
In vivo |
|||||
In vivo Formulation Calculator (Clear solution)
Step 1: Enter information below (Recommended: An additional animal making an allowance for loss during the experiment)
Step 2: Enter the in vivo formulation (This is only the calculator, not formulation. Please contact us first if there is no in vivo formulation at the solubility Section.)
Calculation results:
Working concentration: mg/ml;
Method for preparing DMSO master liquid: mg drug pre-dissolved in μL DMSO ( Master liquid concentration mg/mL, Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug. )
Method for preparing in vivo formulation: Take μL DMSO master liquid, next addμL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O, mix and clarify.
Method for preparing in vivo formulation: Take μL DMSO master liquid, next add μL Corn oil, mix and clarify.
Note: 1. Please make sure the liquid is clear before adding the next solvent.
2. Be sure to add the solvent(s) in order. You must ensure that the solution obtained, in the previous addition, is a clear solution before proceeding to add the next solvent. Physical methods such
as vortex, ultrasound or hot water bath can be used to aid dissolving.
Chemical Information, Storage & Stability
| Molecular Weight | 632.65 | Formula | C34H34F2N4O6 |
Storage (From the date of receipt) | |
|---|---|---|---|---|---|
| CAS No. | 849217-64-7 | Download SDF | Storage of Stock Solutions |
|
|
| Synonyms | GSK1363089, EXEL-2880, XL-880, GSK089 | Smiles | COC1=CC2=C(C=CN=C2C=C1OCCCN3CCOCC3)OC4=C(C=C(C=C4)NC(=O)C5(CC5)C(=O)NC6=CC=C(C=C6)F)F | ||
Mechanism of Action
| Targets/IC50/Ki |
Met
(Cell-free assay) 0.4 nM
KDR
(Cell-free assay) 0.86 nM
Tie-2
(Cell-free assay) 1.1 nM
VEGFR3/FLT4
(Cell-free assay) 2.8 nM
RON
(Cell-free assay) 3 nM
FLT3
(Cell-free assay) 3.6 nM
PDGFRα
(Cell-free assay) 3.6 nM
Kit
(Cell-free assay) 6.7 nM
VEGFR1/FLT1
(Cell-free assay) 6.8 nM
PDGFRβ
(Cell-free assay) 9.6 nM
|
|---|---|
| In vitro |
XL880 inhibits HGF receptor family tyrosine kinases with IC50 values of 0.4 nM for Met and 3 nM for Ron. XL880 also inhibits KDR, Flt-1, and Flt-4 with IC50 values of 0.9 nM, 6.8 nM and 2.8 nM, respectively. XL880 inhibits colony growth of B16F10, A549 and HT29 cells with IC50 of 40 nM, 29 nM and 165 nM, respectively. A recent study indicates XL880 affects cell growth differently in gastric cancer cell lines MKN-45 and KATO-III. XL880 inhibits phosphorylation of MET and downstream signaling molecules in MKN-45 cells, while targets GFGR2 in KATO-III cells.
|
| Kinase Assay |
Kinase Inhibition Assay
|
|
Kinase inhibition is investigated using one of three assay formats: [33P]phosphoryl transfer, luciferase-coupled chemiluminescence, or AlphaScreen tyrosine kinase technology. IC50s are calculated by nonlinear regression analysis using XLFit.33P -Phosphoryl Transfer Kinase Assay Reactions are performed in 384-well white, clear bottom, high-binding microtiter plates (Greiner, Monroe, NC). Plates are coated with 2 μg/well of protein or peptide substrate in a 50 μL volume of coating buffer contained 40 μg/mL substrate (poly(Glu, Tyr) 4:1, 22.5 mM Na2CO3, 27.5 mM NaHCO3, 50 mM NaCl and 3 mM NaN 3. Coated plates are washed once with 50 μL of assay buffer following overnight incubation at room temperature (RT). Test compounds and enzymes are combined with 33P-γ-ATP (3.3 μCi/nmol) in a total volume of 20 μL. The reaction mixture is incubated at RT for 2 hours and terminated by aspiration. The microtiter plates are subsequently washed 6 times with 0.05% Tween-PBS buffer (PBST). Scintillation fluid (50 μL/well) is added and incorporated 33P is measured by liquid scintillation spectrometry using a MicroBeta scintillation counter.Luciferase-Coupled Chemiluminescence Assay Reactions are conducted in 384-well white, medium binding microtiter plates (Greiner). In a first step enzyme and compound are combined and incubated for 60 minutes; reactions are initiated by addition of ATP and peptide substrate (poly(Glu, Tyr) 4:1) in a final voume of 20 μL, and incubated at RT for 2-4 hours. Following the kinase reaction, a 20 μL aliquot of Kinase Glo (Promega, Madison, WI) is added and luminescence signal is measured using a Victor plate reader. Total ATP consumption is limited to 50%. AlphaScreenTM Tyrosine Kinase Assay Donor beads coated with streptavidin and acceptor beads coated with PY100 anti-phosphotyrosine antibody are used. Biotinylated poly(Glu,Tyr) 4:1 is used as the substrate. Substrate phosphorylation is measured by addition of donor/acceptor beads by luminescence following donor-acceptor bead complex formation. Kinase and test compounds are combined and preincubated for 60 minutes, followed by addition of ATP, and biotinylated poly(Glu, Tyr) in a total volume of 20 μL in 384-well white, medium binding microtiter plates (Greiner). Reaction mixtures are incubated for 1 hour at room temperature. Reactions are quenched by addition of 10 μL of 15-30 μg/mL AlphaScreen bead suspension containing 75 mM Hepes, pH 7.4, 300 mM NaCl, 120 mM EDTA, 0.3% BSA and 0.03% Tween-20. After 2-16 hours incubation at room temperature plates are read using an AlphaQuest reader.
|
|
| In vivo |
A single 100 mg/kg oral gavage dose of XL880 results in substantial inhibition of phosphorylation of B16F10 tumor Met and ligand (e.g., HGFor VEGF)-induced receptor phosphorylation of Met in liver and Flk-1/KDR in lung, which both persisted through 24 hours. Treatment with XL880 (30-100 mg/kg, once daily, oral gavage) results in reduction in tumor burden. The lung surface tumor burden is reduced by 50% and 58% following treatment with 30 and 100 mg/kg XL880, respectively. XL880 treatment of mice bearing B16F10 solid tumors also results in dose-dependent tumor growth inhibition of 64% and 87% at 30 and 100 mg/kg, respectively. For both studies, administration of XL880 is well tolerated with no significant body weight loss. XL880 is developed to target abnormal signaling of HGF through Met and simultaneously target several receptors tyrosine kinase involved in tumor angiogenesis. XL880 caused tumor hemorrhage and necrosis in human xenografts within 2 to 4 hours, and maximal tumornecrosis is observed at 96 hours (after five daily doses), resulting in complete regression.
|
References |
|
Applications
| Methods | Biomarkers | Images | PMID |
|---|---|---|---|
| Western blot | p-Met / Met / p-Akt / Akt p-MDM2 / p53 / PUMA pROS1 / tROS1 / pSHP2 / pERK / ERK |
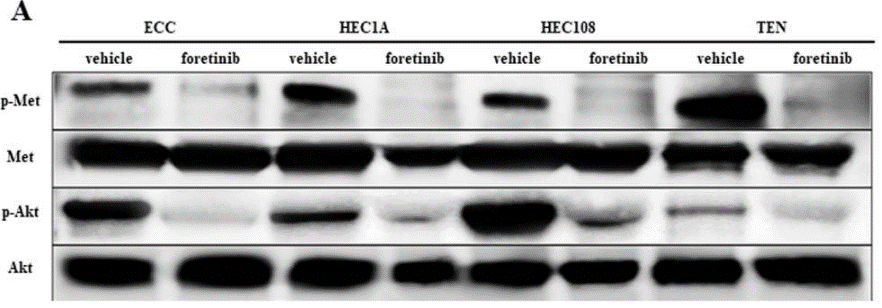
|
29854314 |
| Growth inhibition assay | Cell viability |
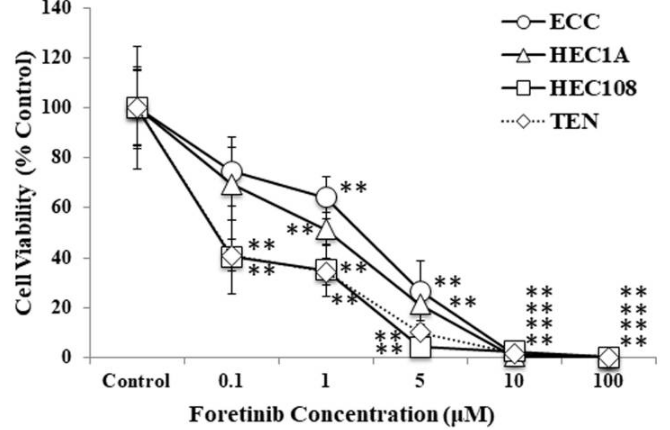
|
29854314 |
Clinical Trial Information
(data from https://clinicaltrials.gov, updated on 2024-05-22)
| NCT Number | Recruitment | Conditions | Sponsor/Collaborators | Start Date | Phases |
|---|---|---|---|---|---|
| NCT00920192 | Completed | Carcinoma Hepatocellular |
GlaxoSmithKline |
August 12 2009 | Phase 1 |
| NCT00742261 | Completed | Solid Tumours |
GlaxoSmithKline |
August 11 2008 | Phase 1 |
| NCT00725764 | Completed | Neoplasms Head and Neck |
GlaxoSmithKline |
August 27 2007 | Phase 2 |
| NCT00725712 | Completed | Neoplasms Gastrointestinal Tract |
GlaxoSmithKline |
March 31 2007 | Phase 2 |
| NCT00743067 | Completed | Solid Tumours |
GlaxoSmithKline |
August 9 2006 | Phase 1 |
Tech Support
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
Signaling Pathway Map
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.