-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
research use only
JNJ-38877605 c-Met inhibitor
Cat.No.S1114
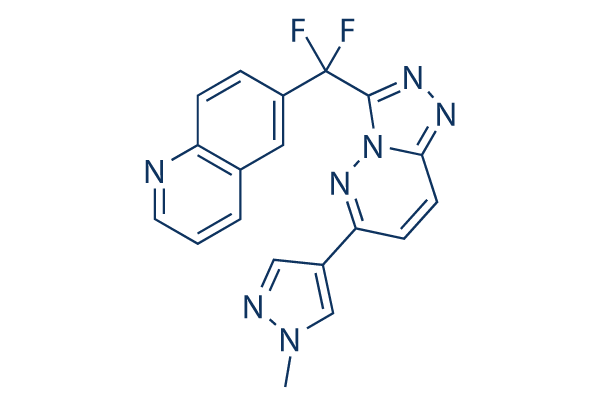
Chemical Structure
Molecular Weight: 377.35
Quality Control
| Related Targets | EGFR VEGFR PDGFR FGFR Src MEK CSF-1R FLT3 HER2 c-Kit |
|---|---|
| Other c-Met Inhibitors | Tepotinib Dihexa SGX-523 Foretinib PHA-665752 SU11274 BMS-777607 Tivantinib PF-04217903 Savolitinib (AZD6094) |
Cell Culture, Treatment & Working Concentration
| Cell Lines | Assay Type | Concentration | Incubation Time | Formulation | Activity Description | PMID |
|---|---|---|---|---|---|---|
| human EBC1 cells | Proliferation assay | 72 h | Antiproliferative activity against human EBC1 cells after 72 hrs, IC50=9.5 nM | |||
| human MKN45 cells | Proliferation assay | 72 h | Antiproliferative activity against human MKN45 cells after 72 hrs, IC50=10.9 nM | |||
| human SNU5 cells | Proliferation assay | 72 h | Antiproliferative activity against human SNU5 cells after 72 hrs, IC50=15.8 nM | |||
| mouse BAF3/TPR-Met cells | Proliferation assay | 72 h | Antiproliferative activity against mouse BAF3/TPR-Met cells after 72 hrs, IC50=17.6 nM | |||
| Click to View More Cell Line Experimental Data | ||||||
Solubility
|
In vitro |
DMSO
: Insoluble
Water : Insoluble Ethanol : Insoluble |
Molarity Calculator
|
In vivo |
|||||
In vivo Formulation Calculator (Clear solution)
Step 1: Enter information below (Recommended: An additional animal making an allowance for loss during the experiment)
Step 2: Enter the in vivo formulation (This is only the calculator, not formulation. Please contact us first if there is no in vivo formulation at the solubility Section.)
Calculation results:
Working concentration: mg/ml;
Method for preparing DMSO master liquid: mg drug pre-dissolved in μL DMSO ( Master liquid concentration mg/mL, Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug. )
Method for preparing in vivo formulation: Take μL DMSO master liquid, next addμL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O, mix and clarify.
Method for preparing in vivo formulation: Take μL DMSO master liquid, next add μL Corn oil, mix and clarify.
Note: 1. Please make sure the liquid is clear before adding the next solvent.
2. Be sure to add the solvent(s) in order. You must ensure that the solution obtained, in the previous addition, is a clear solution before proceeding to add the next solvent. Physical methods such
as vortex, ultrasound or hot water bath can be used to aid dissolving.
Chemical Information, Storage & Stability
| Molecular Weight | 377.35 | Formula | C19H13F2N7 |
Storage (From the date of receipt) | |
|---|---|---|---|---|---|
| CAS No. | 943540-75-8 | Download SDF | Storage of Stock Solutions |
|
|
| Synonyms | N/A | Smiles | CN1C=C(C=N1)C2=NN3C(=NN=C3C(C4=CC5=C(C=C4)N=CC=C5)(F)F)C=C2 | ||
Mechanism of Action
| Targets/IC50/Ki |
c-Met
4 nM
|
|---|---|
| In vitro |
JNJ-38877605 shows more than 600-fold selectivity for c-Met compared with more than 200 other diverse tyrosine and serine-threonine kinases and also potently inhibits HGF-stimulated and constitutively activated c-Met phosphorylation in vitro. In EBC1, GTL16, NCI-H1993, and MKN45 cells, this compound (500 nM) leads to a significant reduction of phosphorylation of Met and RON, another key player in invasive growth. A recent study shows that this chemical is involved in modulating secretion of IL-8, GROa, uPAR and IL-6 in GTL16 cells.
|
| In vivo |
In mice bearing established GTL16 xenografts, JNJ-38877605, dosed orally with 40 mg/kg/day for 72 hours, results in a statistically significant decrease in the plasma levels of human IL-8 (from 0.150 ng/mL to 0.050 ng/mL) and GROα (from 0.080 ng/mL to 0.030 ng/mL). While concentrations of uPAR in the blood become reduced to more than 50% at the same dose.
|
References |
|
Applications
| Methods | Biomarkers | Images | PMID |
|---|---|---|---|
| Western blot | p-MET / HSP27 / p-HSP27 / HSP70 / HSP90 p-EGFR / EGFR / p-FAK / FAK / p-AKT / AKT / p-ERK / ERK |
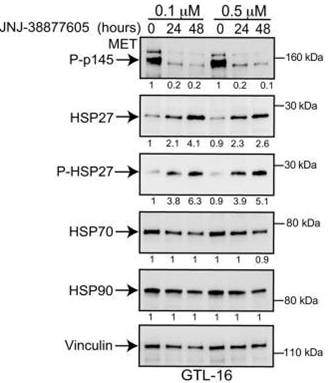
|
24903273 |
Clinical Trial Information
(data from https://clinicaltrials.gov, updated on 2024-05-22)
| NCT Number | Recruitment | Conditions | Sponsor/Collaborators | Start Date | Phases |
|---|---|---|---|---|---|
| NCT00651365 | Terminated | Neoplasms |
Johnson & Johnson Pharmaceutical Research & Development L.L.C.|Ortho Biotech Inc. |
February 2008 | Phase 1 |
Tech Support
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
Signaling Pathway Map
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.