Caspases are the family of cysteine aspartate-specific proteases that play a critical role in proteolytic execution, with significant roles in apoptosis, necrosis, cytokine maturation, inflammatory responses, cellular differentiation, and development.
-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
Caspase Inhibitors/Activators
Other Apoptosis Related Inhibitors
Isoform-selective Products
| Cat.No. | Product Name | Information | Product Use Citations | Product Validations |
|---|---|---|---|---|
| S7775 | Emricasan (IDN-6556) | Emricasan (IDN-6556, PF 03491390, PF-03491390) is a potent irreversible pan-caspase inhibitor. Emricasan is an inhibitor of Zika virus infection. |
|
|
| S7023 | Z-VAD-FMK | Z-VAD-FMK (Z-VAD(OMe)-FMK) is a cell-permeable, irreversible pan-caspase inhibitor, blocks all features of apoptosis in THP.1 and Jurkat T-cells. |
|
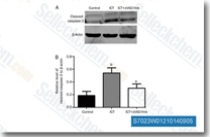
|
| S7311 | Q-VD-Oph | Q-VD-Oph (Quinoline-Val-Asp-Difluorophenoxymethylketone) is a potent pan-caspase inhibitor with IC50 ranged from 25 to 400 nM for caspases 1,3,8, and 9. Q-VD-OPh can inhibits HIV infection. |
|
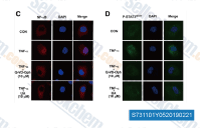
|
| S7312 | Z-DEVD-FMK | Z-DEVD-FMK (Caspase-3 Inhibitor) is a specific, irreversible Caspase-3 inhibitor, and also shows potent inhibition on caspase-6, caspase-7, caspase-8, and caspase-10. |
|

|
| S1029 | CC-5013 (Lenalidomide) | Lenalidomide is a TNF-α secretion inhibitor with IC50 of 13 nM in PBMCs. Lenalidomide (CC-5013) is a ligand of ubiquitin E3 ligase cereblon (CRBN), and it causes selective ubiquitination and degradation of two lymphoid transcription factors, IKZF1 and IKZF3, by the CRBN-CRL4 ubiquitin ligase. Lenalidomide promotes cleaved caspase-3 expression and inhibit VEGF expression and induces apoptosis. |
|
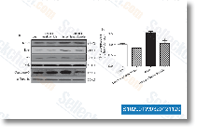
|
| S9042 | Wedelolactone | Wedelolactone, a medicinal plant-derived natural compound, is an inhibitor of IKK that is critical for activation of NF-κB by mediating phosphorylation and degradation of IκBα. This compound is also an inhibitor of caspase-11. |
|
|
| S2768 | Dinaciclib (SCH 727965) | Dinaciclib is a novel and potent CDK inhibitor for CDK2, CDK5, CDK1 and CDK9 with IC50 of 1 nM, 1 nM, 3 nM and 4 nM in cell-free assays, respectively. It also blocks thymidine (dThd) DNA incorporation. Dinaciclib induces apoptosis through the activation of caspases 8 and 9. Phase 3. |
|
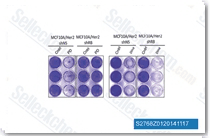
|
| S2228 | Belnacasan (VX-765) | VX-765 (Belnacasan) is a potent and selective inhibitor of caspase-1 with Ki of 0.8 nM in a cell-free assay, and it has reached Phase 2. |
|
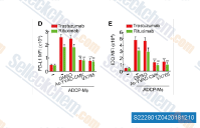
|
| S7314 | Z-IETD-FMK | Z-IETD-FMK (Caspase-8 Inhibitor, Z-IE(OMe)TD(OMe)-FMK) is a specific Caspase-8 inhibitor. Z-IETD-FMK is also an inhibitor of granzyme B. |
|
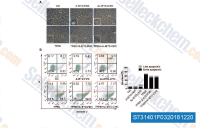
|
| S7901 | Ac-DEVD-CHO | Ac-DEVD-CHO (Caspase-3 Inhibitor I, N-Ac-Asp-Glu-Val-Asp-CHO) is a potent aldehyde inhibitor of Group II caspases with Ki values of 0.2 nM and 0.3 nM for for caspase-3 and caspase-7, respectively. Weak inhibition for caspase-2. |
|
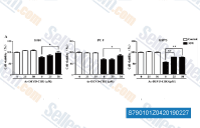
|
Signaling Pathway Map
Caspases can be traditionally divided into two groups based on their sequence homology and function. Caspase-1, -4 and -5 belong to Group I (inflammatory) caspases (caspase-1-related subfamily) that are involved in cytokine maturation and the innate immunity. The Group II caspases (caspase-3-related subgroup) are involved in the regulation of apoptosis, which are further divided into two types: initiators (apical caspases) that includes caspase-2, -8, -9, and -10, and effectors (executing caspases) such as caspase-3, -6 and 7. These caspases have distinct substrate cleavage specificities. To date, over 600 substrates for the cell death-related caspases have been identified. Effector caspases are constitutively produced in cells as dimmers, and the proteolytic processing into cleaved caspases by an initiator enzyme is required to trigger their activity. Being active, effector caspases target a wide spectrum of cellular proteins with the ultimate effect of causing cell death. In contrast to effector caspases, initiator caspases are translated as monomeric zymogens. Formation of multicomponent complexes triggers initiator caspase dimerization sufficient for their activation. Recently, a novel alternative perspective is proposed that mammalian caspases are activated, not to kill, but to extinguish the pro-inflammatory properties of dying cells. This perspective unifies the mammalian caspase family as either positive or negative regulators of inflammation. [1][2]
Caspase-1 subfamily members (caspase-1, -4, and -5) have been implicated as regulators of inflammation through processing and activating two related cytokines, IL-1β and IL-18. The initiator caspases are activated by upstream molecules through protein-protein interaction domains known as caspase recruitment domain (CARD) and death effector domain (DED). The death-inducing signaling complex (DISC), the Apaf-1 apoptosome and the p53-induced protein with a death domain (PIDD) are protein assembly platforms that can recruit caspase-8/-10, -9 and -2, respectively, confirming the essential roles of caspases in both the extrinsic receptor-mediated and intrinsic mitochondrial apoptosis pathways. Caspases are regulated at a post-translational level by inhibitors of apoptosis and by dominant negative isoforms. The proteolytic activity of mature caspase-9 and -3 are subdued by the inhibitor of apoptosis proteins (IAPs). In turn, IAPs are inactivated and caspase activity restored by proteins, such as SMAC/Diablo or HtrA2/Omi, which are released from the mitochondria. The cellular FLICE inhibitory protein (c-FLIP) is a catalytically inactive homologue of caspase-8 and -10, which can prevent their activation by obstructing binding sites on the DISC. Hematopoietic stem cells express a smaller variant, caspase-8L, which acts as a dominant negative when recruited to DISC after CD95 triggering, thereby disrupting the link between CD95 and the caspase cascade. The pro-domain-only polypeptides of caspase-10 have been reported to be pro-apoptotic in some experimental systems, but appeared to be antiapoptotic and capable of inducing NF-κB activity in others. Moreover, either oncogenes (Myc) or tumor suppressors (p53) are able to adjust the intrinsic or the extrinsic caspase cascade involved in the complex signaling system controlling apoptosis. [1][2]
Cells do not necessarily undergo caspase-independent cell death in the absence of active caspases, but may instead survive insult and even promote clonogenic tumor growth. The loss of even one caspase-2 allele results in increased cell proliferation as well as accelerated tumorogenesis, and the loss of caspase-2 expression has been observed in gastric cancer. In a screen of primary breast tumor samples, approximately 75% of the tumors as well as morphologically normal peritumoral tissue samples lack caspase-3 transcripts and caspase-3 protein expression. Reduction of caspase-8 expression has been found in pediatric tumors, and colorectal, gastric, or hepatocellular cancers, as well as in clinical glioma samples. Downregulation of the mitochondrial IAP antagonist Smac/Diablo is associated with renal cell carcinomas, and overexpression of Survivin, another IAP, has been observed in most transformed cell lines and cancers. Caspase-3 and caspase-6 cleavage of Tau protein leads to neurofibrillary tangle formation during Alzheimer’s disease pathogenesis. Moreover, accumulated caspase-6 cleavage-mediated huntingtin fragments represent an early pathological change in the brains of Huntington’s disease patients. Conversely, an increase in cell death is associated with heart disease, stroke, neurodegenerative disorders and liver disease. Additionally, abnormal fluctuations in cytokine levels as a result of the inflammatory response have been implicated in several diseases, including osteoarthritis (OA) and rheumatoid arthritis (RA), gout, inflammatory bowel disorders, sepsis, and inflammatory skin diseases. Thus, the disturbances in the regulation of caspase activation are central for the avoidance of cell death, and have been implicated in the pathogenesis of many disorders including stroke, Alzheimer's disease, myocardial infarction, cancer, and inflammatory disease, which stimulates interest in caspases as potential therapeutic targets. Pralnacasan and VX-765 are reversible caspase-1 inhibitors that are developed for the treatment of a variety of inflammatory disorders disorders, including RA and OA. There is a large pool of inactive procaspase-3 in some cancer cells compared with normal cells, thus, targeting procaspase-3 directly with small molecule activators such as PAC-1 and 1541, rather than targeting upstream regulators of apoptosis could lead to a more effective and direct therapy as caspase-3 is the terminal protease in the apoptotic cascade. [1][3]
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.