
- Bioactive Compounds
- By Signaling Pathways
- PI3K/Akt/mTOR
- Epigenetics
- Methylation
- Immunology & Inflammation
- Protein Tyrosine Kinase
- Angiogenesis
- Apoptosis
- Autophagy
- ER stress & UPR
- JAK/STAT
- MAPK
- Cytoskeletal Signaling
- Cell Cycle
- TGF-beta/Smad
- Compound Libraries
- Antibodies
- Bioreagents
- qPCR
- 2x SYBR Green qPCR Master Mix
- 2x SYBR Green qPCR Master Mix(Low ROX)
- 2x SYBR Green qPCR Master Mix(High ROX)
- Protein Assay
- Protein A/G Magnetic Beads for IP
- Anti-Flag magnetic beads
- Anti-Flag Affinity Gel
- Anti-Myc magnetic beads
- Anti-HA magnetic beads
- Poly FLAG Peptide lyophilized powder
- Protease Inhibitor Cocktail
- Protease Inhibitor Cocktail (EDTA-Free, 100X in DMSO)
- Phosphatase Inhibitor Cocktail (2 Tubes, 100X)
- Cell Biology
- Cell Counting Kit-8 (CCK-8)
- Animal Experiment
- Mouse Direct PCR Kit (For Genotyping)
- New Products
- Contact Us
-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
-
 Other Countries
Other Countries
Bcl-2
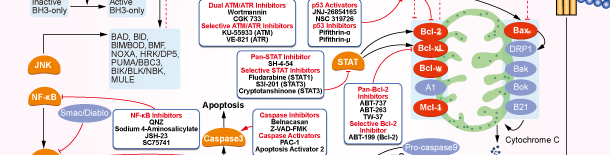
Bcl-2 Products
- All (57)
- Bcl-2 Inhibitors (38)
- Bcl-2 Activators (8)
- Bcl-2 Antagonists (5)
- Bcl-2 Modulators (5)
- New Bcl-2 Products
| Catalog No. | Product Name | Information | Product Use Citations | Product Validations |
|---|---|---|---|---|
| S8048 | Venetoclax (ABT-199) | Venetoclax (ABT-199, GDC-0199) is a Bcl-2-selective inhibitor with Ki of <0.01 nM in cell-free assays, >4800-fold more selective versus Bcl-xL and Bcl-w, and no activity to Mcl-1. Venetoclax is reported to induce cell growth suppression, apoptosis, cell cycle arrest, and autophagy in triple negative breast cancer MDA-MB-231 cells. Phase 3. |
|
|
| S1002 | ABT-737 | ABT-737 is a BH3 mimetic inhibitor of Bcl-xL, Bcl-2 and Bcl-w with EC50 of 78.7 nM, 30.3 nM and 197.8 nM in cell-free assays, respectively; no inhibition observed against Mcl-1, Bcl-B or Bfl-1. ABT-737 induces mitochondrial pathway apoptosis and mitophagy. Phase 2. |

|
|
| S1001 | Navitoclax (ABT-263) | Navitoclax (ABT-263) is a potent inhibitor of Bcl-xL, Bcl-2 and Bcl-w with Ki of ≤ 0.5 nM, ≤1 nM and ≤1 nM in cell-free assays, but binds more weakly to Mcl-1 and A1. Phase 2. |

|
|
| S7747 | Ro-3306 | RO-3306 is an ATP-competitive, and selective CDK1 inhibitor with Ki of 20 nM, >15-fold selectivity against a diverse panel of human kinases. RO-3306 enhances p53-mediated Bax activation and mitochondrial apoptosis. |

|
|
| S1057 | Obatoclax Mesylate (GX15-070) | Obatoclax Mesylate (GX15-070) is an antagonist of Bcl-2 with Ki of 0.22 μM in a cell-free assay, can assist in overcoming MCL-1 mediated resistance to apoptosis. |

|
|
| S8383 | S63845 | S63845 is a new, selective MCL-1 inhibitor with the Kd value of 0.19 nM and has no discernible binding to the other BCL-2 members, BCL-2 or BCL-XL. | ||
| S7790 | A-1210477 | A-1210477 is a potent and selective MCL-1 inhibitor with Ki and IC50 of 0.454 nM and 26.2 nM, respectively, >100-fold selectivity over other Bcl-2 family members. |
|
|
| S1121 | TW-37 | TW-37 is a novel nonpeptide inhibitor to recombinant Bcl-2, Bcl-xL and Mcl-1 with Ki of 0.29 μM, 1.11 μM and 0.26 μM in cell-free assays, respectively. |

|
|
| S7801 | A-1331852 | A-1331852 is a potent and selectiveBCL-XL inhibitor with Ki value less than 0.01 nM for BCL-XL and 6 nM, 4 nM, 142 nM for Bcl-2, Bcl-W, MCL-1 respectively. It may be useful in the treatment of cancer, immune and autoimmune diseases. |
|
|
| S7800 | A-1155463 Dihydrochloride | A-1155463 Dihydrochloride, a highly potent and selective BCL-XL inhibitor, shows picomolar binding affinity to BCL-XL, and >1000-fold weaker binding to BCL-2 and related proteins BCL-W(Ki=19 nM) and MCL-1(Ki>440 nM). |
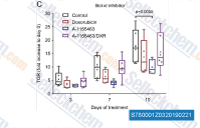
|
|
| S2606 | Mifepristone | Mifepristone is a remarkably active antagonist of progesterone receptor and glucocorticoid receptor with IC50 of 0.2 nM and 2.6 nM, respectively. Mifepristone promotes cell autophagy and apoptosis, decreases Bcl-2 level and increases Beclin1 level, accompanied by weakened interaction between Bcl-2 and Beclin1. |

|
|
| S7531 | UMI-77 | UMI-77 is a selective Mcl-1 inhibitor with Ki of 490 nM, showing selectivity over other members of Bcl-2 family. |

|
|
| S2812 | (R)-(-)-Gossypol (AT-101) acetic acid | (R)-(-)-Gossypol (AT-101) acetic acid, the R-(-) enantiomer of Gossypol acetic acid, binds with Bcl-2, Bcl-xL and Mcl-1 with Ki of 0.32 μM, 0.48 μM and 0.18 μM in cell-free assays; does not inhibit BIR3 domain and BID. AT-101 simultaneously triggers apoptosis and a cytoprotective type of autophagy. Phase 2. |

|
|
| S8643 | AZD5991 | AZD5991 is a macrocyclic MCL-1 inhibitor with sub-nanomolar affinity for MCL-1 (Ki = 0.13 nM). The binding affinity of AZD5991 is about 25-fold lower for mouse Mcl-1 vs. human Mcl-1 but only four-fold lower for rat Mcl-1. | ||
| S8061 | Sabutoclax | Sabutoclax (BI-97C1) is a pan-Bcl-2 inhibitor, including Bcl-xL, Bcl-2, Mcl-1 and Bfl-1 with IC50 of 0.31 μM, 0.32 μM, 0.20 μM and 0.62 μM, respectively. |
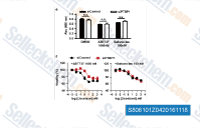
|
|
| S7100 | WEHI-539 | WEHI-539 has high affinity (IC50=1.1 nM) and selectivity for BCL-XL and potently kills cells by selectively antagonizing its prosurvival activity. It has more than a 400-fold higher affinity for BCL-XL versus other prosurvival BCL-2 family members. | ||
| S1071 | HA14-1 | HA14-1 is a non-peptidic ligand of a Bcl-2 surface pocket with IC50 of ~9 μM. |
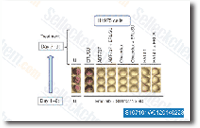
|
|
| S8836 | MIK665 (S64315) | MIK665 (S64315) is an inhibitor of induced myeloid leukemia cell differentiation protein Mcl-1 with Ki value of 1.2 nM and has potential pro-apoptotic and antineoplastic activities. | ||
| S6709 | Obatoclax (GX15-070) | Obatoclax (GX15-070) is an antagonist of Bcl-2 with an Ki of 0.22 μM in a cell-free assay, can assist in overcoming MCL-1 mediated resistance to apoptosis. |
||
| S8820 | Unesbulin (PTC596) | Unesbulin (PTC596) is a second-generation BMI-1 inhibitor that accelerates BMI-1 degradation. PTC596 downregulates MCL-1 and induces p53-independent mitochondrial apoptosis. IC50 values at 72 hours ranged from 68 to 340 nM in mantle cell lymphoma (MCL) cell lines. | ||
| S2448 | Gambogic Acid | Gambogic Acid (Guttatic Acid, Guttic Acid, Beta-Guttiferrin) activates caspases with EC50 of 0.78-1.64 μM and competitively inhibits Bcl-XL, Bcl-2, Bcl-W, Bcl-B, Bfl-1 and Mcl-1 with IC50 of 1.47, 1.21, 2.02, 0.66, 1.06 and 0.79 μM, respectively. | ||
| S7126 | Marinopyrrole A (Maritoclax) | Marinopyrrole A (Maritoclax) is a selective Mcl-1 antagonist. It binds to Mcl-1, but not Bcl-XL, and targets Mcl-1 for proteasomal degradation. Maritoclax disrupts the interaction between Bim and Mcl-1 with an IC50 of 10.1 μM. | ||
| S8865 | BAI1 | BAI1 is a direct allosteric inhibitor of BAX with a dissociation constant (Kd) of 15.0 ± 4 μM. | ||
| S6852 | Gossypol | Gossypol (BL 193) is an orally-active polyphenol isolated from cotton seeds and roots. Gossypol is a potent inhibitor of 5α-reductase 1 and 3α-hydroxysteroid dehydrogenase with IC50 of 3.33 μM and 0.52 μM in cell-free assay, respectively. Gossypol also inhibits the binding of BH3 peptide to Bcl protein with IC50 of 0.4 μM and 10 μM for Bcl-XL and Bcl-2, respectively. Gossypol induces apoptosis and cell growth inhibition in various cancer cells. | ||
| E2926New | A-1155463 | A-1155463 is a highly potent and selective BCL-XL inhibitor with EC50 of 65 nM in H146 cells. | ||
| S8650 | BTSA1 | BTSA1 is a pharmacologically optimized BAX activator that binds with high affinity and specificity to the N-terminal activation site and induces conformational changes to BAX leading to BAX-mediated apoptosis. It effectively promotes apoptosis in leukemia cell lines and patient samples while sparing healthy cells. | ||
| S8759 | S55746 | S55746 (S 055746,BCL201) is a novel, orally active BCL-2 specific inhibitor (Ki = 1.3 nM) with poor affinity for BCL-XL and no significant binding to MCL-1, BFL-1 (BCL2A1/A1). The selectivity of S55746 for BCL-2 versus BCL-XL ranges from ~70 to 400 folds. | ||
| S7105 | BAM7 | BAM 7 is a direct and selective activator of proapoptotic Bax with EC50 of 3.3 μM. | ||
| S3275 | Senkyunolide I | Senkyunolide I (SEI, SENI) is an orally active compound isolated from Ligusticum chuanxiong with analgesic, anti-migraine, neuroprotective, anti-oxidation and anti-apoptosis activities. Senkyunolide I (SEI, SENI) up-regulates the phosphorylation of Erk1/2 and induces Nrf2 nuclear translocation with enhanced HO-1 and NQO1 expressions. Senkyunolide I (SEI, SENI) promotes the ratio of Bcl-2/Bax and inhibits the expressions of cleaved caspase 3 and caspase 9. | ||
| S5967 | Berberine chloride hydrate | Berberine (Natural Yellow 18) chloride hydrate is a quaternary ammonium salt from the group of isoquinoline alkaloids. Berberine activates caspase 3 and caspase 8, cleavage of poly ADP-ribose polymerase (PARP) and the release of cytochrome c. Berberine chloride decreases the expression of c-IAP1, Bcl-2 and Bcl-XL. Berberine chloride induces apoptosis with sustained phosphorylation of JNK and p38 MAPK, as well as generation of the ROS. Berberine chloride is a dual topoisomerase I and II inhibitor. Berberine chloride is also a potential autophagy modulator. | ||
| S8199 | Ruxotemitide (LTX 315) | Ruxotemitide (LTX 315) is the oncolytic peptide that kills cancer cells through Bax/Bak-regulated mitochondrial membrane permeabilization. | ||
| S7849 | BDA-366 | BDA-366 is a small-molecule Bcl2-BH4 domain antagonist and binds BH4 with high affinity and selectivity. It directly binds to Bcl2 with high binding affinity (Ki =3.3 ± 0.73 nM). | ||
| S3238 | Resibufogenin | Resibufogenin (Bufogenin, Recibufogenin), a component of huachansu with anticancer effect, triggers necroptosis through upregulating receptor-interacting protein kinase 3 (RIP3) and phosphorylating mixed lineage kinase domain-like protein at Ser358. Resibufogenin exerts cytotoxic effect by inducing reactive oxygen species (ROS) accumulation. Resibufogenin induces apoptosis and caspase-3 and caspase-8 activity. Resibufogenin increases Bax/Bcl-2 expression, and suppresses cyclin D1, cyclin E, PI3K, p-AKT, p-GSK3β and β-catenin protein expression. | ||
| S0563 | 10-Deacetyl-7-xylosyl paclitaxel | 10-Deacetyl-7-xylosyl paclitaxel (10-Deacetyl-7-xylosyltaxol, 7-xylosyl-10-deacetylpaclitaxel), a derivative of paclitaxel and naturally occurring xyloside isolated from Taxus chinensis, causes significant mitotic arrest in PC-3 cells followed by up-regulating expression of pro-apoptotic Bax and Bad protein, as well as down-regulating expression of anti-apoptotic Bcl-2 and Bcl-XL , which leads to a disturbance of the mitochondrial membrane permeability and to the activation of caspase-9. | ||
| E0124 | Chelerythrine | Chelerythrine (Toddaline, Broussonpapyrine) is a potent, selective antagonist of PKC with an IC50 of 0.66 μM. Chelerythrine also inhibits the BclXL-Bak BH3 peptide binding with an IC50 of 1.5 μM. Chelerythrine shows antitumor, antidiabetic and anti-inflammatory activity. |
||
| E3037 | Solanum lyratum Extract | Solanum lyratum Extract (300 μg/ml) increases Bax levels and decreases Bcl-2 levels, which cause the loss of mitochondrial membrane potential (Δæm) followed by cytochrome C release and caspase-9 and -3 activation, finally leading to apoptosis. Solanum lyratum Extract (300 μg/ml) also promotes p53 and p27, but decreases the levels of cyclin B1 thus causing S-phase arrest. | ||
| S3224 | Cinobufagin | Cinobufagin (Cinobufagine), an active ingredient of Venenum Bufonis, inhibits tumor development. Cinobufagin increases ATM and Chk2 and decreases CDC25C, CDK1, and cyclin B. Cinobufagin inhibits PI3K, AKT and Bcl-2 while increases levels of cleaved caspase-9 and caspase-3. Thus, Cinobufagin induces cell cycle arrest at the G2/M phase and apoptosis. | ||
| S6895 | TCPOBOP | TCPOBOP is a constitutive androstane receptor (CAR) agonist. TCPOBOP attenuates Fas-induced murine liver injury by altering Bcl-2 proteins. | ||
| S9970 | APG-2575 (lisaftoclax) | APG-2575 (lisaftoclax) is a dual Bcl-2 and Bcl-xl inhibitor with IC50 values of 2 nM and 5.9 nM for Bcl-2 and Bcl-xl, respectively. | ||
| S3245 | Nodakenetin | Nodakenetin (NANI), a plant-derived coumarin isolated from Angelica decursiva, inhibits α-glucosidase, PTP1B, rat lens aldose reductase (RLAR), AChE, BChE, and β-site amyloid precursor protein cleaving enzyme 1 (BACE1). Nodakenetin alters the protein expression of Bax and Bcl-2, and prompts mitochondrial apoptosis. Nodakenetin exhibits anti-tumor activity. | ||
| S3267 | Nicotiflorin (Kaempferol-3-O-rutinoside) | Nicotiflorin (Kaempferol-3-O-rutinoside), a flavonoid extracted from Carthamus tinctorius, alters the shape and structure of injured neurons, decreases the number of apoptotic cells, down-regulates expression of p-JAK2, p-STAT3, caspase-3, and Bax and decreases Bax immunoredactivity, and increases Bcl-2 protein expression and immunoreactivity. | ||
| S3243 | Zeaxanthin | Zeaxanthin, the carotenoid alcohol participates in the xanthophyll cycle, activates the extrinsic apoptosis pathway which induces apoptosis on uveal melanoma cells with IC50 value 40.8 µM. | ||
| E2354 | Valepotriate | Valepotriate, an unstable iridoid isolated from Valeriana jatamansi Jones, has anti-epileptic by significantly increasing the expression of GABAA, glutamic acid decarboxylase 65, and Bcl-2 and reduce the expression of caspase-3. | ||
| E2028 | Humanin | Humanin (human) (1-24-Protein humanin (human)), a small mitochondrial-derived cytoprotective polypeptide encoded by mtDNA, exhibits protective effects in several cell types against cellular stress conditions and apoptosis through regulating various signaling mechanisms, such as JAK/STAT pathway and interaction of BCL-2 family of protein. | ||
| E0011 | Linderalactone | Linderalactone inhibits human lung cancer growth by modulating the expression of apoptosis-related proteins (Bax and Bcl-2) with an IC50 of 15 µM in A-549 cells. Linderalactone induces G2/M cell cycle arrest and could also suppress the JAK/STAT signalling pathway. Linderalactone can be isolated from Radix linderae. | ||
| S9276 | Alisol B | Alisol B, a triterpene from Alismatis rhizoma, induces Bax up-regulation and nuclear translocation, the activation of initiator caspase-8 and caspase-9, and executor caspase-3, suggesting the involvement of both extrinsic and intrinsic apoptosis pathways. | ||
| S2271 | Berberine chloride | Berberine chloride is a quaternary ammonium salt from the group of isoquinoline alkaloids. Berberine activates caspase 3 and caspase 8, cleavage of poly ADP-ribose polymerase (PARP) and the release of cytochrome c. Berberine chloride decreases the expression of c-IAP1, Bcl-2 and Bcl-XL. Berberine chloride induces apoptosis with sustained phosphorylation of JNK and p38 MAPK, as well as generation of the ROS. Berberine chloride is a dual topoisomerase I and II inhibitor. Berberine chloride is also a potential autophagy modulator. | ||
| S5600 | Flavokawain A | Flavokawain A, extracted from kava, is an apoptotic inducers and anticarcinogenic agent. Flavokawain A can down-regulation of antiapoptotic proteins, such as XIAP, survivin, and Bcl-xL, thereby changing the balance between apoptotic and antiapoptotic molecules and then induce cell death in tumor cells. | ||
| S8177 | BH3I-1 | BH3I-1 is a Bcl-XL-BH3 domain interaction inhibitor with Ki of 2.4 μM (by fluorescence polarization ).It is a selective inhibitor of Bcl-2 family proteins. | ||
| S8924 | DT2216 | DT2216 is a potent and selective degrader of BCL-XL based on PROTAC technology. DT2216 inhibits various BCL-XL-dependent leukemia and cancer cells but considerably less toxic to platelets. | ||
| S6004 | CCI-007 | CCI-007 is a novel small molecule with cytotoxic activity against infant leukemia with MLL rearrangements. | ||
| S9665 | Motixafortide (BL-8040) | Motixafortide (BL-8040, BKT140, TF 14016, 4-fluorobenzoyl, 4F-benzoyl-TN14003, T140) is an antagonist of CXCR4 with IC50 of ~1 nM. BL-8040 induces the apoptosis of AML blasts by down-regulating ERK, BCL-2, MCL-1 and cyclin-D1 via altered miR-15a/16-1 expression. | ||
| S8758 | VU661013 | VU661013 is a novel, potent, selective MCL1 inhibitor with Ki of 97 ± 30 pM of human MCL-1 in a TR-FRET assay. However, VU661013 does not significantly inhibit BCL-xL or BCL-2 with Ki > 40 μM or = 0.73 μM. VU661013 de-stabilizes BIM/MCL-1 association, leads to apoptosis in AML. | ||
| S5550 | Ethyl gallate | Ethyl gallat (Phyllemblin, gallic acid ethyl ester), which could be found naturally in a variety of plant sources, is a food additive with antimicrobial activity. Ethyl gallat activates the death receptor-dependent pathway of apoptosis by enhancing the expression of caspases-8, -9, and -3 and the Bcl-2 interacting domain (Bid). | ||
| S6990 | PhytohemagglutininPhytohemagglutinin (PHA) | PhytohemagglutininPhytohemagglutinin (PHA) is expressed in Pichia pastoris using native signal peptides, or the Saccharomyces alpha-factor preprosequence, to direct proteins into the secretory pathway. Phytohemagglutinin induces apoptosis in human HEp-2 carcinoma cells via increasing proapoptotic protein Bax and activating caspases-3. | ||
| E2663 | BT2 | BT2 is a novel branched-chain α-ketoacid dehydrogenase complex kinase (BDK) inhibitor wieh an IC50 of 3.19 μM. | ||
| E3020 | Hedyotic Diffusa Extract | Hedyotic Diffusa Extract is extracted from Hedyotic diffusa, of which polysaccharides inhibit the growth of CNE2 cells in a dose-and-time-dependent way, the mechanism may involve induction of cell apoptosis, which is associated with the activation of Bax and caspase-3 protein and the down-regulation of Bcl-2 protein expression. | ||
| S8048 | Venetoclax (ABT-199) | Venetoclax (ABT-199, GDC-0199) is a Bcl-2-selective inhibitor with Ki of <0.01 nM in cell-free assays, >4800-fold more selective versus Bcl-xL and Bcl-w, and no activity to Mcl-1. Venetoclax is reported to induce cell growth suppression, apoptosis, cell cycle arrest, and autophagy in triple negative breast cancer MDA-MB-231 cells. Phase 3. |

|
|
| S1002 | ABT-737 | ABT-737 is a BH3 mimetic inhibitor of Bcl-xL, Bcl-2 and Bcl-w with EC50 of 78.7 nM, 30.3 nM and 197.8 nM in cell-free assays, respectively; no inhibition observed against Mcl-1, Bcl-B or Bfl-1. ABT-737 induces mitochondrial pathway apoptosis and mitophagy. Phase 2. |

|
|
| S1001 | Navitoclax (ABT-263) | Navitoclax (ABT-263) is a potent inhibitor of Bcl-xL, Bcl-2 and Bcl-w with Ki of ≤ 0.5 nM, ≤1 nM and ≤1 nM in cell-free assays, but binds more weakly to Mcl-1 and A1. Phase 2. |

|
|
| S8383 | S63845 | S63845 is a new, selective MCL-1 inhibitor with the Kd value of 0.19 nM and has no discernible binding to the other BCL-2 members, BCL-2 or BCL-XL. | ||
| S7790 | A-1210477 | A-1210477 is a potent and selective MCL-1 inhibitor with Ki and IC50 of 0.454 nM and 26.2 nM, respectively, >100-fold selectivity over other Bcl-2 family members. |

|
|
| S1121 | TW-37 | TW-37 is a novel nonpeptide inhibitor to recombinant Bcl-2, Bcl-xL and Mcl-1 with Ki of 0.29 μM, 1.11 μM and 0.26 μM in cell-free assays, respectively. |

|
|
| S7801 | A-1331852 | A-1331852 is a potent and selectiveBCL-XL inhibitor with Ki value less than 0.01 nM for BCL-XL and 6 nM, 4 nM, 142 nM for Bcl-2, Bcl-W, MCL-1 respectively. It may be useful in the treatment of cancer, immune and autoimmune diseases. |

|
|
| S7800 | A-1155463 Dihydrochloride | A-1155463 Dihydrochloride, a highly potent and selective BCL-XL inhibitor, shows picomolar binding affinity to BCL-XL, and >1000-fold weaker binding to BCL-2 and related proteins BCL-W(Ki=19 nM) and MCL-1(Ki>440 nM). |

|
|
| S2606 | Mifepristone | Mifepristone is a remarkably active antagonist of progesterone receptor and glucocorticoid receptor with IC50 of 0.2 nM and 2.6 nM, respectively. Mifepristone promotes cell autophagy and apoptosis, decreases Bcl-2 level and increases Beclin1 level, accompanied by weakened interaction between Bcl-2 and Beclin1. |

|
|
| S7531 | UMI-77 | UMI-77 is a selective Mcl-1 inhibitor with Ki of 490 nM, showing selectivity over other members of Bcl-2 family. |

|
|
| S2812 | (R)-(-)-Gossypol (AT-101) acetic acid | (R)-(-)-Gossypol (AT-101) acetic acid, the R-(-) enantiomer of Gossypol acetic acid, binds with Bcl-2, Bcl-xL and Mcl-1 with Ki of 0.32 μM, 0.48 μM and 0.18 μM in cell-free assays; does not inhibit BIR3 domain and BID. AT-101 simultaneously triggers apoptosis and a cytoprotective type of autophagy. Phase 2. |

|
|
| S8643 | AZD5991 | AZD5991 is a macrocyclic MCL-1 inhibitor with sub-nanomolar affinity for MCL-1 (Ki = 0.13 nM). The binding affinity of AZD5991 is about 25-fold lower for mouse Mcl-1 vs. human Mcl-1 but only four-fold lower for rat Mcl-1. | ||
| S8061 | Sabutoclax | Sabutoclax (BI-97C1) is a pan-Bcl-2 inhibitor, including Bcl-xL, Bcl-2, Mcl-1 and Bfl-1 with IC50 of 0.31 μM, 0.32 μM, 0.20 μM and 0.62 μM, respectively. |

|
|
| S1071 | HA14-1 | HA14-1 is a non-peptidic ligand of a Bcl-2 surface pocket with IC50 of ~9 μM. |

|
|
| S8836 | MIK665 (S64315) | MIK665 (S64315) is an inhibitor of induced myeloid leukemia cell differentiation protein Mcl-1 with Ki value of 1.2 nM and has potential pro-apoptotic and antineoplastic activities. | ||
| S8820 | Unesbulin (PTC596) | Unesbulin (PTC596) is a second-generation BMI-1 inhibitor that accelerates BMI-1 degradation. PTC596 downregulates MCL-1 and induces p53-independent mitochondrial apoptosis. IC50 values at 72 hours ranged from 68 to 340 nM in mantle cell lymphoma (MCL) cell lines. | ||
| S2448 | Gambogic Acid | Gambogic Acid (Guttatic Acid, Guttic Acid, Beta-Guttiferrin) activates caspases with EC50 of 0.78-1.64 μM and competitively inhibits Bcl-XL, Bcl-2, Bcl-W, Bcl-B, Bfl-1 and Mcl-1 with IC50 of 1.47, 1.21, 2.02, 0.66, 1.06 and 0.79 μM, respectively. | ||
| S8865 | BAI1 | BAI1 is a direct allosteric inhibitor of BAX with a dissociation constant (Kd) of 15.0 ± 4 μM. | ||
| S6852 | Gossypol | Gossypol (BL 193) is an orally-active polyphenol isolated from cotton seeds and roots. Gossypol is a potent inhibitor of 5α-reductase 1 and 3α-hydroxysteroid dehydrogenase with IC50 of 3.33 μM and 0.52 μM in cell-free assay, respectively. Gossypol also inhibits the binding of BH3 peptide to Bcl protein with IC50 of 0.4 μM and 10 μM for Bcl-XL and Bcl-2, respectively. Gossypol induces apoptosis and cell growth inhibition in various cancer cells. | ||
| E2926New | A-1155463 | A-1155463 is a highly potent and selective BCL-XL inhibitor with EC50 of 65 nM in H146 cells. | ||
| S8759 | S55746 | S55746 (S 055746,BCL201) is a novel, orally active BCL-2 specific inhibitor (Ki = 1.3 nM) with poor affinity for BCL-XL and no significant binding to MCL-1, BFL-1 (BCL2A1/A1). The selectivity of S55746 for BCL-2 versus BCL-XL ranges from ~70 to 400 folds. | ||
| S5967 | Berberine chloride hydrate | Berberine (Natural Yellow 18) chloride hydrate is a quaternary ammonium salt from the group of isoquinoline alkaloids. Berberine activates caspase 3 and caspase 8, cleavage of poly ADP-ribose polymerase (PARP) and the release of cytochrome c. Berberine chloride decreases the expression of c-IAP1, Bcl-2 and Bcl-XL. Berberine chloride induces apoptosis with sustained phosphorylation of JNK and p38 MAPK, as well as generation of the ROS. Berberine chloride is a dual topoisomerase I and II inhibitor. Berberine chloride is also a potential autophagy modulator. | ||
| E0124 | Chelerythrine | Chelerythrine (Toddaline, Broussonpapyrine) is a potent, selective antagonist of PKC with an IC50 of 0.66 μM. Chelerythrine also inhibits the BclXL-Bak BH3 peptide binding with an IC50 of 1.5 μM. Chelerythrine shows antitumor, antidiabetic and anti-inflammatory activity. |
||
| S3224 | Cinobufagin | Cinobufagin (Cinobufagine), an active ingredient of Venenum Bufonis, inhibits tumor development. Cinobufagin increases ATM and Chk2 and decreases CDC25C, CDK1, and cyclin B. Cinobufagin inhibits PI3K, AKT and Bcl-2 while increases levels of cleaved caspase-9 and caspase-3. Thus, Cinobufagin induces cell cycle arrest at the G2/M phase and apoptosis. | ||
| S6895 | TCPOBOP | TCPOBOP is a constitutive androstane receptor (CAR) agonist. TCPOBOP attenuates Fas-induced murine liver injury by altering Bcl-2 proteins. | ||
| S9970 | APG-2575 (lisaftoclax) | APG-2575 (lisaftoclax) is a dual Bcl-2 and Bcl-xl inhibitor with IC50 values of 2 nM and 5.9 nM for Bcl-2 and Bcl-xl, respectively. | ||
| S3243 | Zeaxanthin | Zeaxanthin, the carotenoid alcohol participates in the xanthophyll cycle, activates the extrinsic apoptosis pathway which induces apoptosis on uveal melanoma cells with IC50 value 40.8 µM. | ||
| E2354 | Valepotriate | Valepotriate, an unstable iridoid isolated from Valeriana jatamansi Jones, has anti-epileptic by significantly increasing the expression of GABAA, glutamic acid decarboxylase 65, and Bcl-2 and reduce the expression of caspase-3. | ||
| E0011 | Linderalactone | Linderalactone inhibits human lung cancer growth by modulating the expression of apoptosis-related proteins (Bax and Bcl-2) with an IC50 of 15 µM in A-549 cells. Linderalactone induces G2/M cell cycle arrest and could also suppress the JAK/STAT signalling pathway. Linderalactone can be isolated from Radix linderae. | ||
| S2271 | Berberine chloride | Berberine chloride is a quaternary ammonium salt from the group of isoquinoline alkaloids. Berberine activates caspase 3 and caspase 8, cleavage of poly ADP-ribose polymerase (PARP) and the release of cytochrome c. Berberine chloride decreases the expression of c-IAP1, Bcl-2 and Bcl-XL. Berberine chloride induces apoptosis with sustained phosphorylation of JNK and p38 MAPK, as well as generation of the ROS. Berberine chloride is a dual topoisomerase I and II inhibitor. Berberine chloride is also a potential autophagy modulator. | ||
| S5600 | Flavokawain A | Flavokawain A, extracted from kava, is an apoptotic inducers and anticarcinogenic agent. Flavokawain A can down-regulation of antiapoptotic proteins, such as XIAP, survivin, and Bcl-xL, thereby changing the balance between apoptotic and antiapoptotic molecules and then induce cell death in tumor cells. | ||
| S8177 | BH3I-1 | BH3I-1 is a Bcl-XL-BH3 domain interaction inhibitor with Ki of 2.4 μM (by fluorescence polarization ).It is a selective inhibitor of Bcl-2 family proteins. | ||
| S8924 | DT2216 | DT2216 is a potent and selective degrader of BCL-XL based on PROTAC technology. DT2216 inhibits various BCL-XL-dependent leukemia and cancer cells but considerably less toxic to platelets. | ||
| S6004 | CCI-007 | CCI-007 is a novel small molecule with cytotoxic activity against infant leukemia with MLL rearrangements. | ||
| S9665 | Motixafortide (BL-8040) | Motixafortide (BL-8040, BKT140, TF 14016, 4-fluorobenzoyl, 4F-benzoyl-TN14003, T140) is an antagonist of CXCR4 with IC50 of ~1 nM. BL-8040 induces the apoptosis of AML blasts by down-regulating ERK, BCL-2, MCL-1 and cyclin-D1 via altered miR-15a/16-1 expression. | ||
| S8758 | VU661013 | VU661013 is a novel, potent, selective MCL1 inhibitor with Ki of 97 ± 30 pM of human MCL-1 in a TR-FRET assay. However, VU661013 does not significantly inhibit BCL-xL or BCL-2 with Ki > 40 μM or = 0.73 μM. VU661013 de-stabilizes BIM/MCL-1 association, leads to apoptosis in AML. | ||
| E2663 | BT2 | BT2 is a novel branched-chain α-ketoacid dehydrogenase complex kinase (BDK) inhibitor wieh an IC50 of 3.19 μM. | ||
| E3020 | Hedyotic Diffusa Extract | Hedyotic Diffusa Extract is extracted from Hedyotic diffusa, of which polysaccharides inhibit the growth of CNE2 cells in a dose-and-time-dependent way, the mechanism may involve induction of cell apoptosis, which is associated with the activation of Bax and caspase-3 protein and the down-regulation of Bcl-2 protein expression. | ||
| S7747 | Ro-3306 | RO-3306 is an ATP-competitive, and selective CDK1 inhibitor with Ki of 20 nM, >15-fold selectivity against a diverse panel of human kinases. RO-3306 enhances p53-mediated Bax activation and mitochondrial apoptosis. |

|
|
| S8650 | BTSA1 | BTSA1 is a pharmacologically optimized BAX activator that binds with high affinity and specificity to the N-terminal activation site and induces conformational changes to BAX leading to BAX-mediated apoptosis. It effectively promotes apoptosis in leukemia cell lines and patient samples while sparing healthy cells. | ||
| S7105 | BAM7 | BAM 7 is a direct and selective activator of proapoptotic Bax with EC50 of 3.3 μM. | ||
| S3275 | Senkyunolide I | Senkyunolide I (SEI, SENI) is an orally active compound isolated from Ligusticum chuanxiong with analgesic, anti-migraine, neuroprotective, anti-oxidation and anti-apoptosis activities. Senkyunolide I (SEI, SENI) up-regulates the phosphorylation of Erk1/2 and induces Nrf2 nuclear translocation with enhanced HO-1 and NQO1 expressions. Senkyunolide I (SEI, SENI) promotes the ratio of Bcl-2/Bax and inhibits the expressions of cleaved caspase 3 and caspase 9. | ||
| S3238 | Resibufogenin | Resibufogenin (Bufogenin, Recibufogenin), a component of huachansu with anticancer effect, triggers necroptosis through upregulating receptor-interacting protein kinase 3 (RIP3) and phosphorylating mixed lineage kinase domain-like protein at Ser358. Resibufogenin exerts cytotoxic effect by inducing reactive oxygen species (ROS) accumulation. Resibufogenin induces apoptosis and caspase-3 and caspase-8 activity. Resibufogenin increases Bax/Bcl-2 expression, and suppresses cyclin D1, cyclin E, PI3K, p-AKT, p-GSK3β and β-catenin protein expression. | ||
| S9276 | Alisol B | Alisol B, a triterpene from Alismatis rhizoma, induces Bax up-regulation and nuclear translocation, the activation of initiator caspase-8 and caspase-9, and executor caspase-3, suggesting the involvement of both extrinsic and intrinsic apoptosis pathways. | ||
| S5550 | Ethyl gallate | Ethyl gallat (Phyllemblin, gallic acid ethyl ester), which could be found naturally in a variety of plant sources, is a food additive with antimicrobial activity. Ethyl gallat activates the death receptor-dependent pathway of apoptosis by enhancing the expression of caspases-8, -9, and -3 and the Bcl-2 interacting domain (Bid). | ||
| S6990 | PhytohemagglutininPhytohemagglutinin (PHA) | PhytohemagglutininPhytohemagglutinin (PHA) is expressed in Pichia pastoris using native signal peptides, or the Saccharomyces alpha-factor preprosequence, to direct proteins into the secretory pathway. Phytohemagglutinin induces apoptosis in human HEp-2 carcinoma cells via increasing proapoptotic protein Bax and activating caspases-3. | ||
| S1057 | Obatoclax Mesylate (GX15-070) | Obatoclax Mesylate (GX15-070) is an antagonist of Bcl-2 with Ki of 0.22 μM in a cell-free assay, can assist in overcoming MCL-1 mediated resistance to apoptosis. |

|
|
| S7100 | WEHI-539 | WEHI-539 has high affinity (IC50=1.1 nM) and selectivity for BCL-XL and potently kills cells by selectively antagonizing its prosurvival activity. It has more than a 400-fold higher affinity for BCL-XL versus other prosurvival BCL-2 family members. | ||
| S6709 | Obatoclax (GX15-070) | Obatoclax (GX15-070) is an antagonist of Bcl-2 with an Ki of 0.22 μM in a cell-free assay, can assist in overcoming MCL-1 mediated resistance to apoptosis. |
||
| S7126 | Marinopyrrole A (Maritoclax) | Marinopyrrole A (Maritoclax) is a selective Mcl-1 antagonist. It binds to Mcl-1, but not Bcl-XL, and targets Mcl-1 for proteasomal degradation. Maritoclax disrupts the interaction between Bim and Mcl-1 with an IC50 of 10.1 μM. | ||
| S7849 | BDA-366 | BDA-366 is a small-molecule Bcl2-BH4 domain antagonist and binds BH4 with high affinity and selectivity. It directly binds to Bcl2 with high binding affinity (Ki =3.3 ± 0.73 nM). | ||
| S8199 | Ruxotemitide (LTX 315) | Ruxotemitide (LTX 315) is the oncolytic peptide that kills cancer cells through Bax/Bak-regulated mitochondrial membrane permeabilization. | ||
| S0563 | 10-Deacetyl-7-xylosyl paclitaxel | 10-Deacetyl-7-xylosyl paclitaxel (10-Deacetyl-7-xylosyltaxol, 7-xylosyl-10-deacetylpaclitaxel), a derivative of paclitaxel and naturally occurring xyloside isolated from Taxus chinensis, causes significant mitotic arrest in PC-3 cells followed by up-regulating expression of pro-apoptotic Bax and Bad protein, as well as down-regulating expression of anti-apoptotic Bcl-2 and Bcl-XL , which leads to a disturbance of the mitochondrial membrane permeability and to the activation of caspase-9. | ||
| S3245 | Nodakenetin | Nodakenetin (NANI), a plant-derived coumarin isolated from Angelica decursiva, inhibits α-glucosidase, PTP1B, rat lens aldose reductase (RLAR), AChE, BChE, and β-site amyloid precursor protein cleaving enzyme 1 (BACE1). Nodakenetin alters the protein expression of Bax and Bcl-2, and prompts mitochondrial apoptosis. Nodakenetin exhibits anti-tumor activity. | ||
| S3267 | Nicotiflorin (Kaempferol-3-O-rutinoside) | Nicotiflorin (Kaempferol-3-O-rutinoside), a flavonoid extracted from Carthamus tinctorius, alters the shape and structure of injured neurons, decreases the number of apoptotic cells, down-regulates expression of p-JAK2, p-STAT3, caspase-3, and Bax and decreases Bax immunoredactivity, and increases Bcl-2 protein expression and immunoreactivity. | ||
| E2028 | Humanin | Humanin (human) (1-24-Protein humanin (human)), a small mitochondrial-derived cytoprotective polypeptide encoded by mtDNA, exhibits protective effects in several cell types against cellular stress conditions and apoptosis through regulating various signaling mechanisms, such as JAK/STAT pathway and interaction of BCL-2 family of protein. | ||
| E2926New | A-1155463 | A-1155463 is a highly potent and selective BCL-XL inhibitor with EC50 of 65 nM in H146 cells. |
Choose Selective Bcl-2 Inhibitors
Tags: Bcl-2 apoptosis | Bcl-2 inhibition | Bcl-2 protein | Bax apoptosis | Bak protein | Bax protein | Bcl-2 cancer | Bcl-2 expression | Bcl-2 lymphoma | Bcl-2 protein family | Bak apoptosis | Bcl-2 pathway | Bcl-2 phosphorylation | Mcl-1 apoptosis | Bcl-xL apoptosis | Bcl-2 apoptosis pathway | Bcl-2 activation | Bcl-xL protein | Bcl-2 cleavage | Bcl-2 inhibitor clinical trial | Bcl-2 inhibitor review
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.