ATM/ATR inhibitors have emerged as promising therapeutic agents in cancer research, targeting the key kinases of the DNA damage response (DDR) pathway to disrupt the survival of cancer cells with inherent genomic instability. The ATM (ataxia-telangiectasia mutated) and ATR (ataxia-telangiectasia and Rad3-related) kinases are central regulators of DDR, orchestrating a complex network of signaling cascades that sense DNA damage, activate repair mechanisms, and maintain genomic integrity. In cancer cells, which often harbor increased DNA damage due to oncogenic stress and defective repair pathways, the inhibition of ATM/ATR kinases can induce synthetic lethality, selectively eliminating cancer cells while sparing normal cells.
-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
ATM/ATR inhibitors
| Cat.No. | Product Name | Information | Product Use Citations | Product Validations |
|---|---|---|---|---|
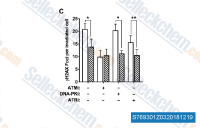
| S7693 | Ceralasertib (AZD6738) | Ceralasertib (AZD6738) is an orally active and selective ATR kinase inhibitor with IC50 of 1 nM, currently in Phase 1/2 clinical trials. |
|
|
| S8680 | AZD1390 | AZD1390 is a first-in-class orally available and CNS penetrant ATM inhibitor with an IC50 of 0.78 nM in cells and >10,000-fold selectivity over closely related members of the PIKK family of enzymes and excellent selectivity across a broad panel of kinases. |
|
|
| S7102 | Berzosertib (VE-822) | Berzosertib (VE-822, VX970, M6620) is an ATR inhibitor with IC50 of 19 nM in HT29 cells. |
|

|
| S2817 | Torin 2 | Torin 2 is a potent and selective mTOR inhibitor with IC50 of 0.25 nM in p53−/− MEFs cell line; 800-fold greater selectivity for mTOR than PI3K and improved pharmacokinetic properties. This compound inhibits ATM/ATR/DNA-PK with EC50 of 28 nM/35 nM/118 nM,in PC3 cell lines respectively. It decreases cell viability and induces autophagy and apoptosis. |
|

|
| E1057 | Lartesertib (M4076) | Lartesertib (M4076) inhibits Ataxia telangiectasia-mutated (ATM) kinase activity with a sub-nanomolar potency and shows remarkable selectivity against other protein kinases, suppresses DSB repair, clonogenic cancer cell growth, and potentiates antitumor activity of ionizing radiation in cancer cell lines. |
|
|
| E1108 | Camonsertib (RP-3500) | Camonsertib (RP-3500) is a highly potent and selective inhibitor of ATR kinase with an IC50 of 1 nM. | ||
| S1570 | KU-60019 | KU-60019 is an improved analogue of KU-55933, with IC50 of 6.3 nM for ATM in cell-free assays, 270- and 1600-fold more selective for ATM than DNA-PK and ATR,and is a highly effective radiosensitizer. |
|

|
| S1092 | KU-55933 | KU-55933 is a potent and specific ATM inhibitor with IC50/Ki of 12.9 nM/2.2 nM in cell-free assays, and is highly selective for ATM as compared to DNA-PK, PI3K/PI4K, ATR and mTOR. KU‑55933 (ATM Kinase Inhibitor) inhibits the activation of autophagy‑initiating kinase ULK1 and results in a significant decrease of autophagy. |
|

|
| S8007 | VE-821 | VE-821(ATR inhibitor IV) is a potent and selective ATP competitive inhibitor of ATR with Ki/IC50 of 13 nM/26 nM in cell-free assays, shows inhibition of H2AX phosphorylation, minimal activity against PIKKs ATM, DNA-PK, mTOR and PI3Kγ. |
|

|
| S4157 | Chloroquine diphosphate | Chloroquine diphosphate is a 4-aminoquinoline anti-malarial and anti-rheumatoid agent, also acting as an ATM activator. Chloroquine is also an inhibitor of toll-like receptors (TLRs). |
|

|
Signaling Pathway Map
ATM/ATR Kinase Biology: Core Regulators of DNA Damage Response Pathway
The ATM and ATR kinases belong to the phosphatidylinositol 3-kinase-related kinase (PIKK) family, sharing structural similarities but differing in their activation triggers and downstream substrates. ATM is primarily activated by double-strand breaks (DSBs) in DNA, a severe form of DNA damage induced by ionizing radiation (IR), chemotherapeutic agents, or replication stress. Upon activation, ATM undergoes autophosphorylation and dimer dissociation, enabling it to phosphorylate a wide range of downstream targets involved in DSB repair, cell cycle checkpoint control, and apoptosis. In contrast, ATR is activated by single-strand DNA (ssDNA) intermediates, which arise during DNA replication stress, UV-induced damage, or incomplete DSB repair. ATR forms a complex with ATRIP (ATR-interacting protein) at ssDNA-coated RPA (replication protein A), initiating a signaling cascade that regulates the S-phase and G2/M checkpoints to prevent the propagation of damaged DNA.
The DNA Damage Response Pathway: Coordination of ATM/ATR Signaling
The DNA damage response pathway is a highly coordinated network that integrates ATM/ATR signaling with DNA repair mechanisms, cell cycle checkpoints, and transcriptional regulation. Upon DNA damage, ATM/ATR activation leads to the phosphorylation of key checkpoint kinases, including Chk2 (by ATM) and Chk1 (by ATR). These kinases then phosphorylate downstream effectors such as Cdc25 phosphatases, which inhibit cyclin-dependent kinases (CDKs) to arrest the cell cycle at G1/S, S, or G2/M checkpoints. This cell cycle arrest provides time for DNA repair: ATM promotes homologous recombination (HR) and non-homologous end joining (NHEJ) for DSB repair, while ATR facilitates nucleotide excision repair (NER) and translesion synthesis (TLS) for ssDNA damage. Additionally, ATM/ATR signaling modulates transcriptional responses through the phosphorylation of transcription factors, including p53, which plays a critical role in determining cell fate after DNA damage.
p53-Mediated Cell Fate Decisions in ATM/ATR Signaling
p53 is a pivotal tumor suppressor gene that acts as a downstream target of both ATM and ATR kinases, integrating DNA damage signals to regulate cell cycle arrest, DNA repair, or apoptosis. ATM directly phosphorylates p53 at serine 15, a modification that stabilizes p53 by inhibiting its interaction with MDM2, an E3 ubiquitin ligase that promotes p53 degradation. ATR also phosphorylates p53 at serine 15, albeit indirectly through Chk1, enhancing p53 stability and transcriptional activity. Activated p53 binds to specific DNA response elements, transactivating target genes such as p21 (a CDK inhibitor that induces cell cycle arrest) and Bax (a pro-apoptotic protein that triggers mitochondrial-mediated apoptosis). The interplay between ATM/ATR and p53 is critical for maintaining genomic stability: in normal cells, this interaction ensures that mild DNA damage is repaired, while severe damage induces apoptosis. However, in many cancers, p53 is mutated or functionally inactivated, disrupting this regulatory loop and contributing to genomic instability and therapeutic resistance.
ATM/ATR Inhibitors: Design, Mechanisms, and Preclinical Research
The critical role of ATM/ATR kinases in cancer cell survival has driven the development of small-molecule inhibitors targeting these kinases. ATM/ATR inhibitors are designed to bind to the kinase domain of ATM or ATR, blocking their catalytic activity and disrupting DDR signaling. These inhibitors can be classified into two categories: selective inhibitors (targeting either ATM or ATR) and dual inhibitors (targeting both kinases). Selective ATM inhibitors, such as KU-55933 and AZD0156, have been shown to sensitize cancer cells to IR and DSB-inducing chemotherapeutics by abrogating DSB repair and cell cycle checkpoints. Selective ATR inhibitors, including VE-821 and AZD6738, are particularly effective in cancers with high replication stress, such as those harboring mutations in BRCA1/2, TP53, or MYC. Dual ATM/ATR inhibitors, such as M4344, offer the potential to target multiple DDR pathways, enhancing synthetic lethality in a broader range of cancer types.
Mechanisms of Synthetic Lethality in ATM/ATR Inhibitor-Treated Cancer Cells
Synthetic lethality is a key mechanism underlying the efficacy of ATM/ATR inhibitors in cancer therapy, referring to the phenomenon where the combination of two genetic or pharmacological perturbations results in cell death, while each perturbation alone is non-lethal. In cancer cells with defective DDR pathways (e.g., BRCA1/2 mutations, p53 inactivation), the inhibition of ATM/ATR kinases creates a "synthetic lethal" interaction by eliminating the remaining DNA repair and checkpoint mechanisms. For example, BRCA1/2-deficient cancer cells rely on ATM-mediated HR for DSB repair; thus, ATM inhibition blocks HR, leading to the accumulation of unrepaired DSBs and cell death. Similarly, p53-mutant cancer cells are more sensitive to ATR inhibitors because they lack the p53-mediated G1 checkpoint, relying solely on ATR-dependent S and G2/M checkpoints to survive replication stress. Inhibiting ATR in these cells abrogates these checkpoints, resulting in catastrophic DNA damage and mitotic catastrophe.
Preclinical Evaluation of ATM/ATR Inhibitors in Cancer Models
Preclinical studies have demonstrated the efficacy of ATM/ATR inhibitors in a wide range of cancer models, including breast, ovarian, lung, and colorectal cancers. In BRCA1/2-mutant breast and ovarian cancer xenografts, treatment with ATM inhibitors (e.g., AZD0156) significantly enhances the anti-tumor activity of IR and platinum-based chemotherapeutics. ATR inhibitors (e.g., AZD6738) have shown promising results in preclinical models of triple-negative breast cancer (TNBC), which is characterized by high replication stress and frequent p53 mutations. Additionally, combination therapy with ATM/ATR inhibitors and immune checkpoint inhibitors (e.g., anti-PD-1/PD-L1 antibodies) has emerged as a potential strategy to enhance anti-tumor immunity. Preclinical data suggest that ATM/ATR inhibition induces immunogenic cell death (ICD) by promoting the release of damage-associated molecular patterns (DAMPs), such as calreticulin and ATP, which activate dendritic cells and enhance T-cell infiltration into the tumor microenvironment.
Translational Research and Clinical Challenges of ATM/ATR Inhibitors
Despite the promising preclinical results, the translation of ATM/ATR inhibitors into clinical practice faces several challenges, including therapeutic resistance, toxicity to normal cells, and the identification of predictive biomarkers. To address these challenges, ongoing translational research is focused on understanding the mechanisms of resistance to ATM/ATR inhibitors, developing combination therapies to overcome resistance, and identifying biomarkers that can select patients most likely to benefit from treatment. Biomarker discovery is particularly critical, as the efficacy of ATM/ATR inhibitors is highly dependent on the genetic background of the tumor. Potential biomarkers include mutations in DDR genes (e.g., BRCA1/2, TP53, ATM, ATR), gene expression signatures of replication stress, and the presence of genomic instability (e.g., high tumor mutational burden, chromosomal instability).
Clinical Trials of ATM/ATR Inhibitors in Cancer Patients
Several ATM/ATR inhibitors are currently undergoing clinical trials for the treatment of various cancers, either as monotherapy or in combination with other anti-cancer agents. For example, AZD6738 (a selective ATR inhibitor) is being evaluated in phase II trials for the treatment of TNBC, non-small cell lung cancer (NSCLC), and ovarian cancer, in combination with chemotherapy or radiation therapy. AZD0156 (a selective ATM inhibitor) is in phase I/II trials for the treatment of advanced solid tumors, including BRCA1/2-mutant cancers, in combination with olaparib (a PARP inhibitor) or radiation therapy. Early clinical data suggest that ATM/ATR inhibitors are well-tolerated, with manageable toxicities such as fatigue, nausea, and myelosuppression. However, further clinical studies are needed to determine the optimal dose, scheduling, and combination strategies to maximize anti-tumor efficacy while minimizing toxicity.
Future Directions in ATM/ATR Inhibitor Research
Future research on ATM/ATR inhibitors will focus on several key areas: (1) Elucidating the molecular mechanisms of resistance to ATM/ATR inhibitors, including the activation of alternative DDR pathways and the acquisition of mutations in ATM/ATR or their downstream targets; (2) Developing novel combination therapies, such as combining ATM/ATR inhibitors with PARP inhibitors, immune checkpoint inhibitors, or targeted therapies (e.g., CDK4/6 inhibitors) to enhance synthetic lethality and overcome resistance; (3) Identifying and validating predictive biomarkers to guide patient selection and personalize treatment; (4) Improving the selectivity of ATM/ATR inhibitors to reduce toxicity to normal cells, particularly hematopoietic and gastrointestinal cells, which are highly sensitive to DDR inhibition. Additionally, exploring the role of ATM/ATR inhibitors in other diseases characterized by genomic instability, such as neurodegenerative disorders and aging, may expand their therapeutic applications beyond cancer.
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.