-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
research use only
BGJ398 (Infigratinib) FGFR inhibitor
Cat.No.S2183
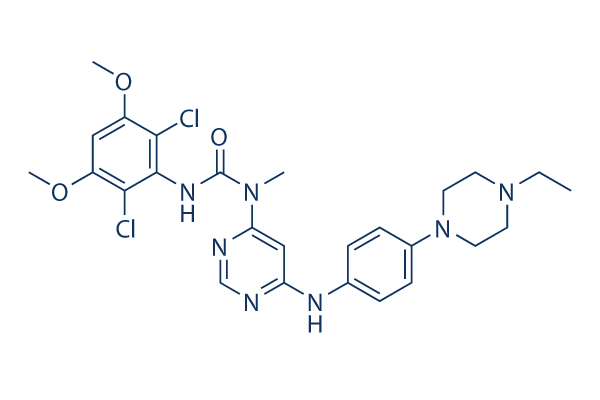
Chemical Structure
Molecular Weight: 560.48
Quality Control
Products Often Used Together with BGJ398 (Infigratinib)
| Related Targets | EGFR VEGFR JAK PDGFR Src HIF FLT3 FLT HER2 Bcr-Abl |
|---|---|
| Other FGFR Inhibitors | PD173074 AZD4547 (Fexagratinib) BLU9931 Futibatinib (TAS-120) LY2874455 H3B-6527 PD-166866 Zoligratinib (Debio-1347) Fisogatinib (BLU-554) SSR128129E |
Cell Culture, Treatment & Working Concentration
| Cell Lines | Assay Type | Concentration | Incubation Time | Formulation | Activity Description | PMID |
|---|---|---|---|---|---|---|
| HCC | Growth Inhibition Assay | 1-2500 nM | 48 h | IC50= 2359 nm | 25688743 | |
| HCC | Growth Inhibition Assay | 1-2500 nM | 48 h | IC50=1124 nm | 25688743 | |
| HCT116 | Growth Inhibition Assay | 48 h | IC50=3 μM | 24503538 | ||
| HKH2 | Growth Inhibition Assay | 48 h | IC50=4 μM | 24503538 | ||
| RKO | Growth Inhibition Assay | 48 h | IC50=1.2 μM | 24503538 | ||
| LS174T | Growth Inhibition Assay | 48 h | IC50=4 μM | 24503538 | ||
| HCD9 | Growth Inhibition Assay | 0.5-5 μM | 48/72 h | DMSO | decreases cell viability | 24135816 |
| HCT116 | Growth Inhibition Assay | 0.5-5 μM | 48/72 h | DMSO | decreases cell viability | 24135816 |
| SNU-C1 | Growth Inhibition Assay | 0.5-5 μM | 48/72 h | DMSO | no effect | 24135816 |
| MFE280 | Growth Inhibition Assay | IC50=2.63 ± 0.82 μM | 23443805 | |||
| AN3CA | Growth Inhibition Assay | IC50=1.00 ± 0.20 μM | 23443805 | |||
| HEC155 | Growth Inhibition Assay | IC50=4.74 ± 1.09 μM | 23443805 | |||
| MFE296 | Growth Inhibition Assay | IC50=2.86 ± 0.20 μM | 23443805 | |||
| SPAC1S | Growth Inhibition Assay | IC50=3.19 ± 0.93 μM | 23443805 | |||
| RL952 | Growth Inhibition Assay | IC50=3.41 ± 0.23 μM | 23443805 | |||
| EN1 | Growth Inhibition Assay | IC50=4.75 ± 0.62 μM | 23443805 | |||
| SNGII | Growth Inhibition Assay | IC50=4.29 ± 0.58 μM | 23443805 | |||
| ISHIKAWA | Growth Inhibition Assay | IC50=5.48 ± 0.03 μM | 23443805 | |||
| HEC1A | Growth Inhibition Assay | IC50=10.00 ± 1.00 μM | 23443805 | |||
| KLE | Growth Inhibition Assay | IC50=3.03 ± 0.11 μM | 23443805 | |||
| SNGM | Growth Inhibition Assay | IC50=5.00 ± 0.41 μM | 23443805 | |||
| USPC2 | Growth Inhibition Assay | IC50=7.00 ± 0.21 μM | 23443805 | |||
| EN | Growth Inhibition Assay | IC50=6.03 ± 0.31 μM | 23443805 | |||
| MFE319 | Growth Inhibition Assay | IC50=5.37 ± 0.03 μM | 23443805 | |||
| EFE184 | Growth Inhibition Assay | IC50=8.04 ± 0.69 μM | 23443805 | |||
| ECC1 | Growth Inhibition Assay | IC50=6.74 ± 0.59 μM | 23443805 | |||
| HEC1B | Growth Inhibition Assay | IC50=6.45 ± 0.67 μM | 23443805 | |||
| USPC1 | Growth Inhibition Assay | IC50=5.75 ± 0.50 μM | 23443805 | |||
| SPAC1L | Growth Inhibition Assay | IC50=4.92 ± 0.50 μM | 23443805 | |||
| HCC827 | Function assay | Displacement of [3H]-cyclopamine from SMO V404M mutant in gefitinib resistant human HCC827 cells by scintillation counting, Ki = 0.0465 μM. | 28787156 | |||
| SJ-GBM2 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SJ-GBM2 cells | 29435139 | |||
| NB-EBc1 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for NB-EBc1 cells | 29435139 | |||
| sf9 | Function assay | 60 mins | Inhibition of non-phosphorylated N-terminal His6-tagged FGFR4 C552A mutant (G442 to E753 residues) (unknown origin) expressed in sf9 cells using 5-Fluo-Ahx-KKKKEEIYFFFG-NH2 as substrate after 60 mins by microfluidic mobility shift assay, IC50 = 0.00082 μM. | ChEMBL | ||
| sf9 | Function assay | 60 mins | Inhibition of wild type non-phosphorylated N-terminal His6-tagged FGFR4 (G442 to E753 residues) (unknown origin) expressed in sf9 cells using 5-Fluo-Ahx-KKKKEEIYFFFG-NH2 as substrate after 60 mins by microfluidic mobility shift assay, IC50 = 0.062 μM. | ChEMBL | ||
| sf9 | Function assay | 60 mins | Inhibition of non-phosphorylated N-terminal His6-tagged FGFR4 C477A mutant (G442 to E753 residues) (unknown origin) expressed in sf9 cells using 5-Fluo-Ahx-KKKKEEIYFFFG-NH2 as substrate after 60 mins by microfluidic mobility shift assay, IC50 = 0.064 μM. | ChEMBL | ||
| Click to View More Cell Line Experimental Data | ||||||
Solubility
|
In vitro |
DMSO
: 3 mg/mL
(5.35 mM)
Water : Insoluble Ethanol : Insoluble |
Molarity Calculator
|
In vivo |
|||||
In vivo Formulation Calculator (Clear solution)
Step 1: Enter information below (Recommended: An additional animal making an allowance for loss during the experiment)
Step 2: Enter the in vivo formulation (This is only the calculator, not formulation. Please contact us first if there is no in vivo formulation at the solubility Section.)
Calculation results:
Working concentration: mg/ml;
Method for preparing DMSO master liquid: mg drug pre-dissolved in μL DMSO ( Master liquid concentration mg/mL, Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug. )
Method for preparing in vivo formulation: Take μL DMSO master liquid, next addμL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O, mix and clarify.
Method for preparing in vivo formulation: Take μL DMSO master liquid, next add μL Corn oil, mix and clarify.
Note: 1. Please make sure the liquid is clear before adding the next solvent.
2. Be sure to add the solvent(s) in order. You must ensure that the solution obtained, in the previous addition, is a clear solution before proceeding to add the next solvent. Physical methods such
as vortex, ultrasound or hot water bath can be used to aid dissolving.
Chemical Information, Storage & Stability
| Molecular Weight | 560.48 | Formula | C26H31Cl2N7O3 |
Storage (From the date of receipt) | |
|---|---|---|---|---|---|
| CAS No. | 872511-34-7 | Download SDF | Storage of Stock Solutions |
|
|
| Synonyms | NVP-BGJ398 | Smiles | CCN1CCN(CC1)C2=CC=C(C=C2)NC3=CC(=NC=N3)N(C)C(=O)NC4=C(C(=CC(=C4Cl)OC)OC)Cl | ||
Mechanism of Action
| Targets/IC50/Ki |
FGFR1
(Cell-free assay) 0.9 nM
FGFR3
(Cell-free assay) 1.0 nM
FGFR2
(Cell-free assay) 1.4 nM
FGFR3 (K650E)
(Cell-free assay) 4.9 nM
FGFR4
(Cell-free assay) 60 nM
|
|---|---|
| In vitro |
Infigratinib (BGJ398) also prevents VEGFR2 with low potency, with an IC50 of 0.18 μM for inhibiting this target. It suppresses other kinases including ABL, FYN, KIT, LCK, LYN and YES with IC50 values of 2.3 μM, 1.9 μM, 0.75 μM, 2.5 μM, 0.3 μM and 1.1 μM, respectively. At the cellular level, this compound inhibits the proliferation of the FGFR1-, FGFR2-Q, and FGFR3-dependent BaF3 cells with IC50 of 2.9 μM, 2.0 μM and 2 μM, respectively. It interferes with autophosphorylation on specific tyrosine residues including FGFR-WT, FGFR2-WT, FGFR3-K650E, FGFR3-S249C and FGFR4-WT with IC50 of 4.6 nM, 4.9 nM, 5 nM, 5 nM and 168 nM, respectively. Additionally, it suppresses proliferation of the cancer cells with wild-type (WT) FGFR3 overexpression such as RT112, RT4, SW780 and JMSU1 with IC50 of 5 nM, 30 nM, 32 nM and 15 nM, respectively.
|
| Kinase Assay |
Radiometric kinase assay
|
|
The enzymatic kinase activity is assessed by measuring the phosphorylation of a synthetic substrate by the purified GST-fusion FGFR3-K650E kinase domain, in the presence of radiolabeled ATP. Enzyme activities are measured by mixing 10 μL of a 3-fold concentrated solution of Infigratinib (BGJ398) or control with 10 μL of the corresponding substrate mixture (peptidic substrate, ATP and [γ33P]ATP). The reactions are initiated by addition of 10 μL of a 3-fold concentrated solution of the enzyme in assay buffer. The final concentrations of the assay components are as following: 10 ng of GST-FGFR3-K650E, 20 mM Tris-HCl, pH 7.5, 3 mM MnCl2, 3 mM MgCl2, 1 mM DTT, 250 μg/mL PEG 20000, 2 μg/mL poly(EY) 4:1, 1% DMSO and 0.5 μM ATP (γ-[33P]-ATP 0.1 μCi). The assay is carried out according to the filter binding (FB) method in 96-well plates at room temperature for 10 minutes in a final volume of 30 μL including this compound. The enzymatic reactions are stopped by the addition of 20 μL of 125 mM EDTA, and the incorporation of 33P into the polypeptidic substrates is quantified as following: 30 μL of the stopped reaction mixture are transferred onto Immobilon-PVDF membranes previously soaked for 5 minutes with methanol, rinsed with water, soaked for 5 min with 0.5% H3PO4, and mounted on vacuum manifold with disconnected vacuum source. After spotting, vacuum is connected, and each well rinsed with 0.5% H3PO4 (200 μL). Free membranes are removed and ished four times on a shaker with 1% H3PO4 and once with ethanol. Membranes are dried and overlaid with addition of 10 μL/well of a scintillation fluid. The plates are eventually sealed and counted in a microplate scintillation counter. IC50 values are calculated by linear regression analysis of the percentage inhibition of it.
|
|
| In vivo |
In this orthotopic xenograft bladder cancer model, BGJ398 induces tumor growth inhibition and stasis after oral administration for 12 consecutive days at the doses of 10 and 30 mg/kg, respectively. Interestingly, the animals that received this compound exhibit either no body weight loss (10 mg/kg) or 10% body weight gain (30 mg/kg), a further indication of efficacy. RT112 tumor-bearing and female Rowett rats receive a single oral administration of the monophosphate salt of Infigratinib (BGJ398) at the doses of 4.25 and 8.51 mg/kg. It significantly decreases the levels of pFRS2 and pMAPK in a dose-dependent manner. Additionally, it inhibits significantly bFGF-stimulated angiogenesis in a dose-dependent manner. However, it does not impair VEGF-induced blood vessel formation.
|
References |
Applications
| Methods | Biomarkers | Images | PMID |
|---|---|---|---|
| Western blot | pFGFR1 / FGFR1 / pFRS2 / FRS2 / MEK / ERK p-YAP (S127) / YAP Mcl-1 Cyclin D1 / Cyclin A / Cyclin B / γ-H2AX / Cleaved caspase-9 / PARP Snail / Slug / ZEB1 p-FRS2 / FRS2 / p-AKT / AKT / p-ERK / ERK |
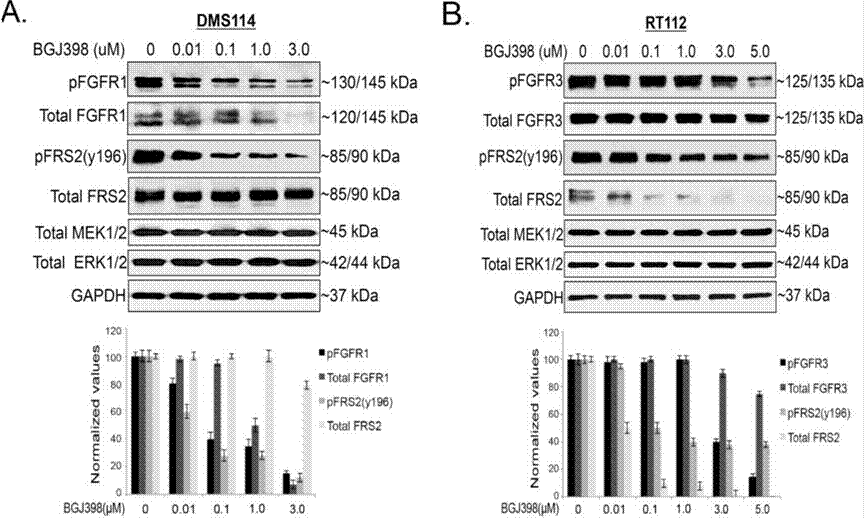
|
28255027 |
| Immunofluorescence | YAP |
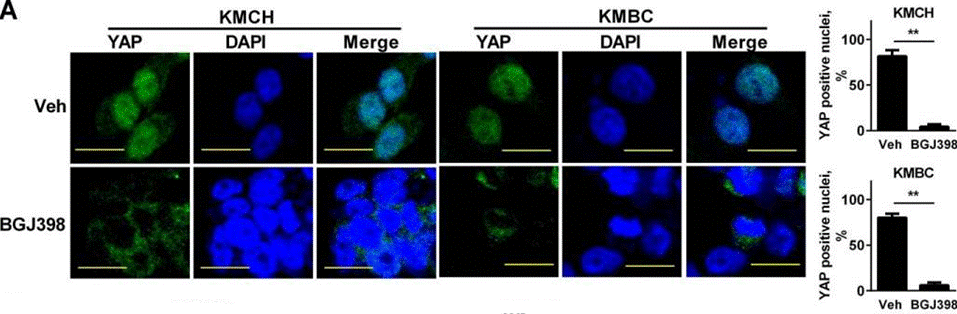
|
26826125 |
| Growth inhibition assay | Cell viability IC50 |
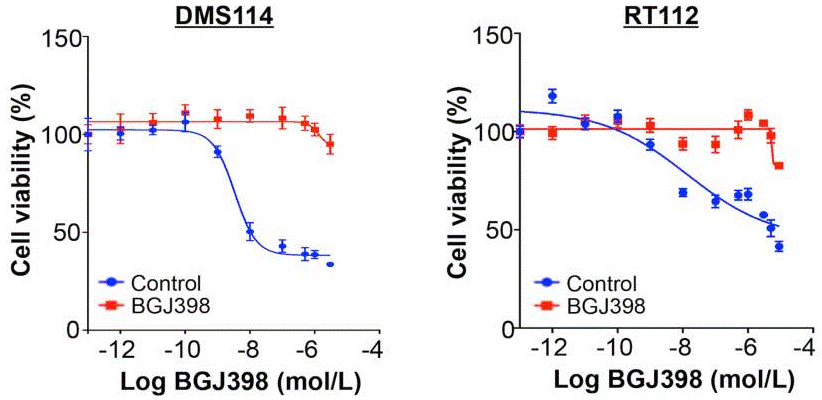
|
28255027 |
Clinical Trial Information
(data from https://clinicaltrials.gov, updated on 2024-05-22)
| NCT Number | Recruitment | Conditions | Sponsor/Collaborators | Start Date | Phases |
|---|---|---|---|---|---|
| NCT03510455 | Terminated | Tumor-Induced Osteomalacia|Oncogenic Osteomalacia |
National Institute of Dental and Craniofacial Research (NIDCR)|National Institutes of Health Clinical Center (CC) |
February 27 2019 | Phase 2 |
| NCT02312804 | Withdrawn | Cancer of Cervix|Tumors |
The University of Texas Health Science Center at San Antonio |
January 2015 | Phase 1 |
| NCT02160041 | Terminated | Solid Tumor|Hematologic Malignancies |
Novartis Pharmaceuticals|Novartis |
July 24 2014 | Phase 2 |
| NCT01928459 | Completed | Advanced Solid Tumors|Metastatic Solid Tumors |
Novartis Pharmaceuticals|Novartis |
October 2013 | Phase 1 |
| NCT01004224 | Completed | Advanced Solid Tumors With Alterations of FGFR1 2 and or 3|Squamous Lung Cancer With FGFR1 Amplification|Bladder Cancer With FGFR3 Mutation or Fusion|Advanced Solid Tumors With FGFR1 Amplication|Advanced Solid Tumors With FGFR2 Amplication|Advanced Solid Tumors With FGFR3 Mutation |
Novartis Pharmaceuticals|Novartis |
December 11 2009 | Phase 1 |
Tech Support
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
Frequently Asked Questions
Question 1:
If you have any suggestions about the formulation of this compound for a direct oral gavage administration?
Answer:
It can be dissolved in 30% PEG400/0.5% Tween80/5% Propylene glycol at 30 mg/ml as a suspension.
Signaling Pathway Map
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.