-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
research use only
PLX-4720 B-Raf Inhibitor
Cat.No.S1152
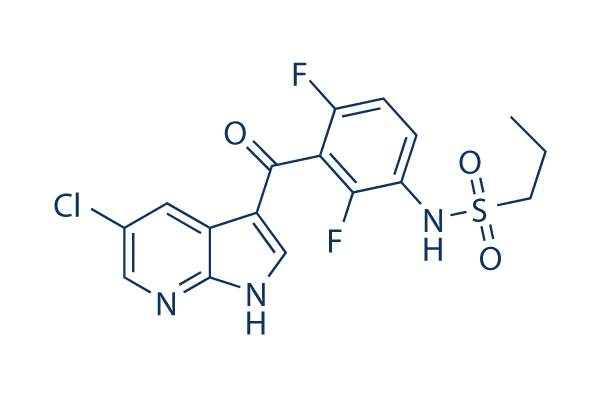
Chemical Structure
Molecular Weight: 413.83
Quality Control
Products Often Used Together with PLX-4720
| Related Targets | ERK p38 MAPK JNK MEK Ras KRas S6 Kinase MAP4K TAK1 Mixed Lineage Kinase |
|---|---|
| Other Raf Inhibitors | LY3009120 Exarafenib (KIN-2787) GDC-0879 Avutometinib (Ro5126766, CH5126766) AZ 628 SB590885 TAK-632 GW5074 RAF265 (CHIR-265) PLX7904 |
Cell Culture, Treatment & Working Concentration
| Cell Lines | Assay Type | Concentration | Incubation Time | Formulation | Activity Description | PMID |
|---|---|---|---|---|---|---|
| DU-4475 | Growth Inhibition Assay | IC50=0.07457 μM | SANGER | |||
| EoL-1-cell | Growth Inhibition Assay | IC50=0.14166 μM | SANGER | |||
| C32 | Growth Inhibition Assay | IC50=0.15131 μM | SANGER | |||
| M14 | Growth Inhibition Assay | IC50=0.21757 μM | SANGER | |||
| CP50-MEL-B | Growth Inhibition Assay | IC50=0.29784 μM | SANGER | |||
| A101D | Growth Inhibition Assay | IC50=0.32589 μM | SANGER | |||
| G-361 | Growth Inhibition Assay | IC50=0.34637 μM | SANGER | |||
| HT-144 | Growth Inhibition Assay | IC50=0.36329 μM | SANGER | |||
| ACN | Growth Inhibition Assay | IC50=0.38477 μM | SANGER | |||
| COLO-829 | Growth Inhibition Assay | IC50=0.38968 μM | SANGER | |||
| MEL-HO | Growth Inhibition Assay | IC50=0.41179 μM | SANGER | |||
| SH-4 | Growth Inhibition Assay | IC50=0.41422 μM | SANGER | |||
| SK-MEL-3 | Growth Inhibition Assay | IC50=0.51568 μM | SANGER | |||
| A375 | Growth Inhibition Assay | IC50=0.67359 μM | SANGER | |||
| MMAC-SF | Growth Inhibition Assay | IC50=0.68614 μM | SANGER | |||
| BHT-101 | Growth Inhibition Assay | IC50=0.70702 μM | SANGER | |||
| K5 | Growth Inhibition Assay | IC50=0.76148 μM | SANGER | |||
| BV-173 | Growth Inhibition Assay | IC50=0.79644 μM | SANGER | |||
| RVH-421 | Growth Inhibition Assay | IC50=0.86796 μM | SANGER | |||
| HCC2218 | Growth Inhibition Assay | IC50=0.87844 μM | SANGER | |||
| WM-115 | Growth Inhibition Assay | IC50=0.88692 μM | SANGER | |||
| SK-MEL-28 | Growth Inhibition Assay | IC50=1.04569 μM | SANGER | |||
| COLO-679 | Growth Inhibition Assay | IC50=1.10464 μM | SANGER | |||
| MZ7-mel | Growth Inhibition Assay | IC50=1.14963 μM | SANGER | |||
| SK-MEL-30 | Growth Inhibition Assay | IC50=1.33386 μM | SANGER | |||
| NCI-H209 | Growth Inhibition Assay | IC50=1.6086 μM | SANGER | |||
| HTC-C3 | Growth Inhibition Assay | IC50=1.66294 μM | SANGER | |||
| KARPAS-45 | Growth Inhibition Assay | IC50=2.04978 μM | SANGER | |||
| NCI-SNU-5 | Growth Inhibition Assay | IC50=2.11969 μM | SANGER | |||
| KP-4 | Growth Inhibition Assay | IC50=2.30787 μM | SANGER | |||
| PA-1 | Growth Inhibition Assay | IC50=2.72673 μM | SANGER | |||
| HuO-3N1 | Growth Inhibition Assay | IC50=2.87946 μM | SANGER | |||
| NCI-H358 | Growth Inhibition Assay | IC50=2.92232 μM | SANGER | |||
| CTB-1 | Growth Inhibition Assay | IC50=3.40176 μM | SANGER | |||
| 697 | Growth Inhibition Assay | IC50=3.55266 μM | SANGER | |||
| CP66-MEL | Growth Inhibition Assay | IC50=4.15927 μM | SANGER | |||
| NB13 | Growth Inhibition Assay | IC50=4.49179 μM | SANGER | |||
| DBTRG-05MG | Growth Inhibition Assay | IC50=4.53325 μM | SANGER | |||
| A2058 | Growth Inhibition Assay | IC50=4.72164 μM | SANGER | |||
| KG-1 | Growth Inhibition Assay | IC50=4.73908 μM | SANGER | |||
| 8305C | Growth Inhibition Assay | IC50=5.1873 μM | SANGER | |||
| RPMI-7951 | Growth Inhibition Assay | IC50=5.80283 μM | SANGER | |||
| CHL-1 | Growth Inhibition Assay | IC50=5.97603 μM | SANGER | |||
| TI-73 | Growth Inhibition Assay | IC50=6.00902 μM | SANGER | |||
| HT-1080 | Growth Inhibition Assay | IC50=6.10946 μM | SANGER | |||
| ES5 | Growth Inhibition Assay | IC50=6.14924 μM | SANGER | |||
| 8-MG-BA | Growth Inhibition Assay | IC50=6.18129 μM | SANGER | |||
| NB7 | Growth Inhibition Assay | IC50=6.21373 μM | SANGER | |||
| H4 | Growth Inhibition Assay | IC50=6.22493 μM | SANGER | |||
| CAL-72 | Growth Inhibition Assay | IC50=6.45423 μM | SANGER | |||
| HCC1806 | Growth Inhibition Assay | IC50=6.81931 μM | SANGER | |||
| BCPAP | Growth Inhibition Assay | IC50=7.21764 μM | SANGER | |||
| LB2241-RCC | Growth Inhibition Assay | IC50=7.36907 μM | SANGER | |||
| COLO-741 | Growth Inhibition Assay | IC50=8.01679 μM | SANGER | |||
| HSC-3 | Growth Inhibition Assay | IC50=8.07068 μM | SANGER | |||
| SW982 | Growth Inhibition Assay | IC50=8.41516 μM | SANGER | |||
| GCT | Growth Inhibition Assay | IC50=8.75314 μM | SANGER | |||
| KY821 | Growth Inhibition Assay | IC50=9.05178 μM | SANGER | |||
| JVM-3 | Growth Inhibition Assay | IC50=9.56999 μM | SANGER | |||
| RS4-11 | Growth Inhibition Assay | IC50=9.6048 μM | SANGER | |||
| VA-ES-BJ | Growth Inhibition Assay | IC50=10.0149 μM | SANGER | |||
| A431 | Growth Inhibition Assay | IC50=10.4212 μM | SANGER | |||
| LXF-289 | Growth Inhibition Assay | IC50=10.458 μM | SANGER | |||
| SK-MEL-24 | Growth Inhibition Assay | IC50=10.8274 μM | SANGER | |||
| NOS-1 | Growth Inhibition Assay | IC50=10.8472 μM | SANGER | |||
| KNS-62 | Growth Inhibition Assay | IC50=11.2404 μM | SANGER | |||
| SK-HEP-1 | Growth Inhibition Assay | IC50=11.3527 μM | SANGER | |||
| A3-KAW | Growth Inhibition Assay | IC50=11.7178 μM | SANGER | |||
| SK-LU-1 | Growth Inhibition Assay | IC50=12.2655 μM | SANGER | |||
| TYK-nu | Growth Inhibition Assay | IC50=12.3932 μM | SANGER | |||
| NMC-G1 | Growth Inhibition Assay | IC50=12.6062 μM | SANGER | |||
| BB65-RCC | Growth Inhibition Assay | IC50=12.7169 μM | SANGER | |||
| QIMR-WIL | Growth Inhibition Assay | IC50=12.8833 μM | SANGER | |||
| D-566MG | Growth Inhibition Assay | IC50=13.9576 μM | SANGER | |||
| KYSE-140 | Growth Inhibition Assay | IC50=14.0753 μM | SANGER | |||
| SCC-4 | Growth Inhibition Assay | IC50=14.3359 μM | SANGER | |||
| U251 | Growth Inhibition Assay | IC50=14.8492 μM | SANGER | |||
| D-542MG | Growth Inhibition Assay | IC50=14.9222 μM | SANGER | |||
| LAMA-84 | Growth Inhibition Assay | IC50=14.9932 μM | SANGER | |||
| NCI-H720 | Growth Inhibition Assay | IC50=15.2684 μM | SANGER | |||
| DEL | Growth Inhibition Assay | IC50=15.4293 μM | SANGER | |||
| SBC-1 | Growth Inhibition Assay | IC50=15.4305 μM | SANGER | |||
| ECC10 | Growth Inhibition Assay | IC50=15.4458 μM | SANGER | |||
| Daoy | Growth Inhibition Assay | IC50=15.7616 μM | SANGER | |||
| SCH | Growth Inhibition Assay | IC50=15.7835 μM | SANGER | |||
| MZ2-MEL | Growth Inhibition Assay | IC50=16.0646 μM | SANGER | |||
| CAL-12T | Growth Inhibition Assay | IC50=16.4862 μM | SANGER | |||
| KE-37 | Growth Inhibition Assay | IC50=16.8107 μM | SANGER | |||
| LS-411N | Growth Inhibition Assay | IC50=17.118 μM | SANGER | |||
| NCI-H2228 | Growth Inhibition Assay | IC50=17.3071 μM | SANGER | |||
| SK-MEL-2 | Growth Inhibition Assay | IC50=17.4965 μM | SANGER | |||
| HN | Growth Inhibition Assay | IC50=17.7248 μM | SANGER | |||
| NCI-H1648 | Growth Inhibition Assay | IC50=17.818 μM | SANGER | |||
| IA-LM | Growth Inhibition Assay | IC50=18.3172 μM | SANGER | |||
| EW-13 | Growth Inhibition Assay | IC50=18.5708 μM | SANGER | |||
| YKG-1 | Growth Inhibition Assay | IC50=19.5711 μM | SANGER | |||
| KNS-81-FD | Growth Inhibition Assay | IC50=19.5858 μM | SANGER | |||
| 23132-87 | Growth Inhibition Assay | IC50=19.7642 μM | SANGER | |||
| NUGC-3 | Growth Inhibition Assay | IC50=19.9887 μM | SANGER | |||
| 5637 | Growth Inhibition Assay | IC50=20.0478 μM | SANGER | |||
| NCI-H1755 | Growth Inhibition Assay | IC50=20.4764 μM | SANGER | |||
| RH-18 | Growth Inhibition Assay | IC50=20.5748 μM | SANGER | |||
| RXF393 | Growth Inhibition Assay | IC50=20.6756 μM | SANGER | |||
| LU-134-A | Growth Inhibition Assay | IC50=20.7056 μM | SANGER | |||
| TE-12 | Growth Inhibition Assay | IC50=20.7201 μM | SANGER | |||
| MOLT-4 | Growth Inhibition Assay | IC50=21.1915 μM | SANGER | |||
| IGR-1 | Growth Inhibition Assay | IC50=21.3796 μM | SANGER | |||
| HOP-92 | Growth Inhibition Assay | IC50=21.4987 μM | SANGER | |||
| SK-MES-1 | Growth Inhibition Assay | IC50=21.7381 μM | SANGER | |||
| LU-65 | Growth Inhibition Assay | IC50=21.8624 μM | SANGER | |||
| MS-1 | Growth Inhibition Assay | IC50=22.1203 μM | SANGER | |||
| LoVo | Growth Inhibition Assay | IC50=22.244 μM | SANGER | |||
| A704 | Growth Inhibition Assay | IC50=22.5155 μM | SANGER | |||
| HT-1376 | Growth Inhibition Assay | IC50=22.6059 μM | SANGER | |||
| IST-MEL1 | Growth Inhibition Assay | IC50=22.6751 μM | SANGER | |||
| Ramos-2G6-4C10 | Growth Inhibition Assay | IC50=22.7366 μM | SANGER | |||
| T47D | Growth Inhibition Assay | IC50=22.7979 μM | SANGER | |||
| HT-1197 | Growth Inhibition Assay | IC50=23.0817 μM | SANGER | |||
| LB2518-MEL | Growth Inhibition Assay | IC50=23.6412 μM | SANGER | |||
| J-RT3-T3-5 | Growth Inhibition Assay | IC50=24.7595 μM | SANGER | |||
| SK-NEP-1 | Growth Inhibition Assay | IC50=24.8744 μM | SANGER | |||
| NCI-H526 | Growth Inhibition Assay | IC50=25.0023 μM | SANGER | |||
| IST-SL1 | Growth Inhibition Assay | IC50=25.2751 μM | SANGER | |||
| HH | Growth Inhibition Assay | IC50=25.3192 μM | SANGER | |||
| NCI-H82 | Growth Inhibition Assay | IC50=25.938 μM | SANGER | |||
| SNU-449 | Growth Inhibition Assay | IC50=27.2018 μM | SANGER | |||
| COR-L23 | Growth Inhibition Assay | IC50=27.2813 μM | SANGER | |||
| LOXIMVI | Growth Inhibition Assay | IC50=27.368 μM | SANGER | |||
| GR-ST | Growth Inhibition Assay | IC50=27.6706 μM | SANGER | |||
| NCI-SNU-1 | Growth Inhibition Assay | IC50=27.944 μM | SANGER | |||
| ALL-PO | Growth Inhibition Assay | IC50=28.1604 μM | SANGER | |||
| ML-2 | Growth Inhibition Assay | IC50=28.2814 μM | SANGER | |||
| HOP-62 | Growth Inhibition Assay | IC50=28.713 μM | SANGER | |||
| EGI-1 | Growth Inhibition Assay | IC50=28.8845 μM | SANGER | |||
| TCCSUP | Growth Inhibition Assay | IC50=28.9272 μM | SANGER | |||
| LB996-RCC | Growth Inhibition Assay | IC50=29.5682 μM | SANGER | |||
| LCLC-97TM1 | Growth Inhibition Assay | IC50=32.1964 μM | SANGER | |||
| NCI-H1304 | Growth Inhibition Assay | IC50=32.3301 μM | SANGER | |||
| KP-N-YS | Growth Inhibition Assay | IC50=32.5973 μM | SANGER | |||
| NCI-H1770 | Growth Inhibition Assay | IC50=33.1648 μM | SANGER | |||
| EM-2 | Growth Inhibition Assay | IC50=33.6504 μM | SANGER | |||
| ChaGo-K-1 | Growth Inhibition Assay | IC50=33.7236 μM | SANGER | |||
| ACHN | Growth Inhibition Assay | IC50=33.8385 μM | SANGER | |||
| MN-60 | Growth Inhibition Assay | IC50=33.8544 μM | SANGER | |||
| EW-18 | Growth Inhibition Assay | IC50=33.8971 μM | SANGER | |||
| KGN | Growth Inhibition Assay | IC50=35.7292 μM | SANGER | |||
| U031 | Growth Inhibition Assay | IC50=35.8132 μM | SANGER | |||
| HMV-II | Growth Inhibition Assay | IC50=36.0774 μM | SANGER | |||
| L-363 | Growth Inhibition Assay | IC50=37.6455 μM | SANGER | |||
| NCI-H1155 | Growth Inhibition Assay | IC50=38.0015 μM | SANGER | |||
| NCI-H1793 | Growth Inhibition Assay | IC50=38.1026 μM | SANGER | |||
| P30-OHK | Growth Inhibition Assay | IC50=38.1332 μM | SANGER | |||
| AN3-CA | Growth Inhibition Assay | IC50=38.1615 μM | SANGER | |||
| UACC-257 | Growth Inhibition Assay | IC50=38.79 μM | SANGER | |||
| MCF7 | Growth Inhibition Assay | IC50=39.8629 μM | SANGER | |||
| KP-N-YN | Growth Inhibition Assay | IC50=40.4285 μM | SANGER | |||
| T98G | Growth Inhibition Assay | IC50=40.4957 μM | SANGER | |||
| HGC-27 | Growth Inhibition Assay | IC50=43.274 μM | SANGER | |||
| NCI-H1092 | Growth Inhibition Assay | IC50=43.2895 μM | SANGER | |||
| KARPAS-299 | Growth Inhibition Assay | IC50=43.3071 μM | SANGER | |||
| LB1047-RCC | Growth Inhibition Assay | IC50=44.9959 μM | SANGER | |||
| 786-0 | Growth Inhibition Assay | IC50=45.65 μM | SANGER | |||
| HCC2157 | Growth Inhibition Assay | IC50=46.0359 μM | SANGER | |||
| NY | Growth Inhibition Assay | IC50=46.1778 μM | SANGER | |||
| EFM-19 | Growth Inhibition Assay | IC50=46.7533 μM | SANGER | |||
| EW-16 | Growth Inhibition Assay | IC50=46.7806 μM | SANGER | |||
| UM-UC-3 | Growth Inhibition Assay | IC50=46.8059 μM | SANGER | |||
| HT-29 | Growth Inhibition Assay | IC50=47.8792 μM | SANGER | |||
| LN-405 | Growth Inhibition Assay | IC50=48.0827 μM | SANGER | |||
| NCI-H727 | Growth Inhibition Assay | IC50=48.7726 μM | SANGER | |||
| D-502MG | Growth Inhibition Assay | IC50=48.9676 μM | SANGER | |||
| GMS-10 | Growth Inhibition Assay | IC50=49.2974 μM | SANGER | |||
| MEL-JUSO | Growth Inhibition Assay | IC50=49.347 μM | SANGER | |||
| insect cells | Function assay | Inhibition of N-terminal His-tagged BRAF V600E mutant (unknown origin) expressed in baculovirus infected insect cells co-expressing CDC37 using biotinylated-MEK as substrate by AlphaScreen assay, IC50 = 0.013 μM. | 29461827 | |||
| A375 | Function assay | Inhibition of BRAF V600E mutant in human A375 cells assessed as reduction in ERK phosphorylation by AlphaScreen assay, IC50 = 0.044 μM. | 29461827 | |||
| A375 | Function assay | Inhibition of b-Raf in human A375 cells assessed phosphorylation of ERK, IC50 = 0.046 μM. | 22808911 | |||
| A375 | Antiproliferative assay | Antiproliferative activity against human A375 cells expressing B-Raf V600E mutant and wild type Ras, IC50 = 0.5 μM. | 22808911 | |||
| A375 | Cytotoxicity assay | 72 hrs | Cytotoxicity against human A375 cells harboring BRAF V600E mutant after 72 hrs by CellTiter-Glo assay, IC50 = 0.5 μM. | 29461827 | ||
| HCT116 | Antiproliferative assay | Antiproliferative activity against human HCT116 cells expressing wild type b-Raf and KRAS mutant, IC50 = 27 μM. | 22808911 | |||
| A673 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for A673 cells | 29435139 | |||
| Saos-2 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for Saos-2 cells | 29435139 | |||
| RD | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for RD cells | 29435139 | |||
| SK-N-SH | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SK-N-SH cells | 29435139 | |||
| NB1643 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for NB1643 cells | 29435139 | |||
| OHS-50 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for OHS-50 cells | 29435139 | |||
| SJ-GBM2 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SJ-GBM2 cells | 29435139 | |||
| SK-N-MC | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SK-N-MC cells | 29435139 | |||
| Click to View More Cell Line Experimental Data | ||||||
Solubility
|
In vitro |
DMSO
: 83 mg/mL
(200.56 mM)
Water : Insoluble Ethanol : Insoluble |
Molarity Calculator
|
In vivo |
|||||
In vivo Formulation Calculator (Clear solution)
Step 1: Enter information below (Recommended: An additional animal making an allowance for loss during the experiment)
Step 2: Enter the in vivo formulation (This is only the calculator, not formulation. Please contact us first if there is no in vivo formulation at the solubility Section.)
Calculation results:
Working concentration: mg/ml;
Method for preparing DMSO master liquid: mg drug pre-dissolved in μL DMSO ( Master liquid concentration mg/mL, Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug. )
Method for preparing in vivo formulation: Take μL DMSO master liquid, next addμL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O, mix and clarify.
Method for preparing in vivo formulation: Take μL DMSO master liquid, next add μL Corn oil, mix and clarify.
Note: 1. Please make sure the liquid is clear before adding the next solvent.
2. Be sure to add the solvent(s) in order. You must ensure that the solution obtained, in the previous addition, is a clear solution before proceeding to add the next solvent. Physical methods such
as vortex, ultrasound or hot water bath can be used to aid dissolving.
Chemical Information, Storage & Stability
| Molecular Weight | 413.83 | Formula | C17H14ClF2N3O3S |
Storage (From the date of receipt) | |
|---|---|---|---|---|---|
| CAS No. | 918505-84-7 | Download SDF | Storage of Stock Solutions |
|
|
| Synonyms | N/A | Smiles | CCCS(=O)(=O)NC1=C(C(=C(C=C1)F)C(=O)C2=CNC3=C2C=C(C=N3)Cl)F | ||
Mechanism of Action
| Targets/IC50/Ki |
C-Raf-1 (Y340D/Y341D)
(Cell-free assay) 6.7 nM
B-Raf (V600E)
(Cell-free assay) 13 nM
BRK
(Cell-free assay) 130 nM
B-Raf
(Cell-free assay) 160 nM
|
|---|---|
| In vitro |
PLX-4720 displays >10 times selectivity against wild type B-Raf, and >100 times selectivity over other kinases such as Frk, Src, Fak, FGFR, and Aurora A with IC50 of 1.3-3.4 μM. This compound significantly inhibits the ERK phosphorylation in cell lines bearing B-RafV600E with IC50 of 14-46 nM, but not the cells with wild-type B-Raf. It significantly inhibits the growth of tumor cell lines bearing the B-RafV600E oncogene, such as COLO205, A375, WM2664, and COLO829 with GI50 of 0.31 μM, 0.50 μM, 1.5 μM, and 1.7 μM, respectively. In addition, this chemical treatment at 1 μM induces cell cycle arrest and apoptosis exclusively in the B-RafV600E-positive 1205Lu cells, but not in the B-Raf wild-type C8161 cells. This compound treatment (10 μM) significantly induces >14-fold expression of BIM in the PTEN+ cells, compared with the PTEN- cell lines (4-fold), giving an explanation of the resistance of PTEN- cells to this chemical-induced apoptosis.
|
| Kinase Assay |
In vitro Raf kinase activities
|
|
The in vitro kinase activities of wild type Raf and mutants are determined by measuring phosphorylation of biotinylated-MEK protein using Perkin-Elmer's AlphaScreen Technology. For each enzyme (0.1 ng), 20-μL reactions are carried out in 20 mM Hepes (pH 7.0), 10 mM MgCl2, 1 mM DTT, 0.01% Tween-20, 100 nM biotin-MEK protein, various ATP concentrations, and increasing concentrations of PLX-4720 at room temperature. Reactions are stopped at 2, 5, 8, 10, 20, and 30 minutes with 5 μL of a solution containing 20 mM Hepes (pH 7.0), 200 mM NaCl, 80 mM EDTA, and 0.3% BSA. The stop solution also includes phospho-MEK Antibody, Streptavidin-coated Donor beads and Protein A Acceptor beads from the AlphaScreen Protein A Detection Kit. The antibody and beads are preincubated in stop solution in the dark at room temperature for 30 minutes. The final dilution of antibody is 1/2,000, and the final concentration of each bead is 10 μg/mL. The assay plates are incubated at room temperature for one hour then are read on a PerkinElmer AlphaQuest reader.
|
|
| In vivo |
Oral administration of PLX-4720 at 20 mg/kg/day induces significant tumor growth delays and regressions in B-RafV600E-dependent COLO205 tumor xenografts, without obvious adverse effects in mice even at dose of 1 g/kg. This compound at 100 mg/kg twice daily almost completely eliminates the 1205Lu xenografts bearing B-RafV600E, while has no activity against C8161 xenografts bearing wild-type B-Raf. The anti-tumor effects of this compound correlate with the blockade of MAPK pathway in those cells harboring the V600E mutation. This chemical treatment at 30 mg/kg/day significant inhibits the tumor growth of 8505c xenografts by >90%, and dramatically decreases distant lung metastases.
|
References |
|
Applications
| Methods | Biomarkers | Images | PMID |
|---|---|---|---|
| Western blot | pAkt(Ser473) / pAkt(Thr308) p27 / Cyclin D1 / pRb p-EGFR 1173 / EGFR / p-Akt / Akt p-MEK / MEK / p-ERK / ERK / p-FAK(S910) |
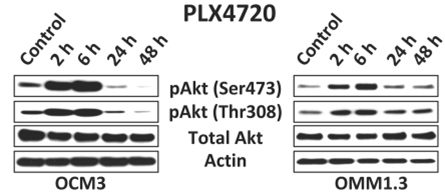
|
21828154 |
| Immunofluorescence | ZKSCAN3 / TFEB LAMP1 |
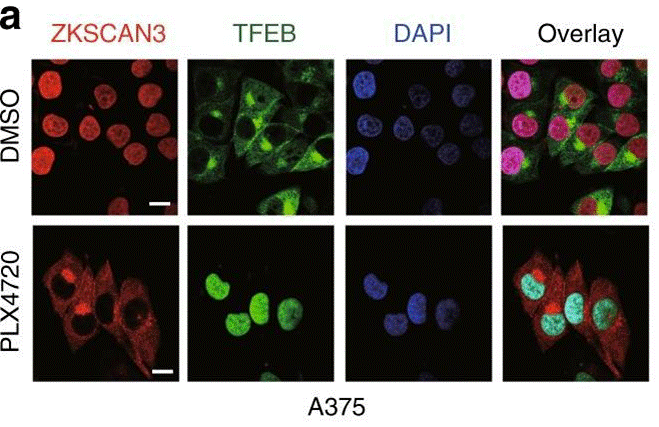
|
30979895 |
| Growth inhibition assay | Cell viability |

|
27848137 |
| ELISA | mIFN-γ |
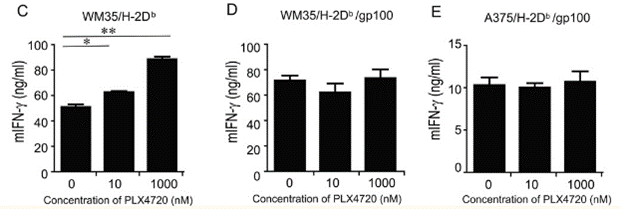
|
23204132 |
Tech Support
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
Frequently Asked Questions
Question 1:
What would you recommend to make working solution for intraperitoneal injection into mice?
Answer:
Due to its very limited solubility in aqueous solution, this compound can be administered as a not fully dissolved suspension via oral gavage. For this reason, we recommend oral gavage for its administration.
Signaling Pathway Map
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.