
- Inhibitors
- By product type
- Natural Products
- Inducing Agents
- Peptides
- Antibiotics
- Antibody-drug Conjugates(ADC)
- PROTAC
- Hydrotropic Agents
- Dyes
- By Signaling Pathways
- PI3K/Akt/mTOR
- Epigenetics
- Methylation
- Immunology & Inflammation
- Protein Tyrosine Kinase
- Angiogenesis
- Apoptosis
- Autophagy
By research - Antibodies
- Compound Libraries
- Bioreagents
- qPCR
- 2x SYBR Green qPCR Master Mix
- 2x SYBR Green qPCR Master Mix(Low ROX)
- 2x SYBR Green qPCR Master Mix(High ROX)
- Protein Assay
- Protein A/G Magnetic Beads for IP
- Anti-Flag magnetic beads
- Anti-Flag Affinity Gel
- Anti-Myc magnetic beads
- Anti-HA magnetic beads
- Poly DYKDDDDK Tag Peptide lyophilized powder
- Protease Inhibitor Cocktail
- Protease Inhibitor Cocktail (EDTA-Free, 100X in DMSO)
- Phosphatase Inhibitor Cocktail (2 Tubes, 100X)
- Cell Biology
- Cell Counting Kit-8 (CCK-8)
- Animal Experiment
- Mouse Direct PCR Kit (For Genotyping)
- Featured Products
- MRTX1133
- Nab-Paclitaxel
- KP-457
- IAG933
- RMC-6236 (Daraxonrasib)
- RMC-7977
- Zoldonrasib (RMC-9805)
- GsMTx4
- Navitoclax (ABT-263)
- TSA (Trichostatin A)
- Y-27632 Dihydrochloride
- SB431542
- SB202190
- MK-2206 Dihydrochloride
- LY294002
- Alisertib (MLN8237)
- XAV-939
- CHIR-99021 (Laduviglusib)
- Bafilomycin A1 (Baf-A1)
- Thiazovivin (TZV)
- CP-673451
- Verteporfin
- DAPT
- Galunisertib (LY2157299)
- MG132
- SBE-β-CD
- Tween 80
- Bavdegalutamide (ARV-110)
- Z-VAD-FMK
- Wnt-C59 (C59)
- IWR-1-endo
- (+)-JQ1
- 3-Deazaneplanocin A (DZNep) Hydrochloride
- RepSox (E-616452)
- Erastin
- Q-VD-Oph
- Puromycin Dihydrochloride
- Cycloheximide
- Telaglenastat (CB-839)
- A-83-01
- Ceralasertib (AZD6738)
- Liproxstatin-1
- Emricasan (IDN-6556)
- PMA (Phorbol 12-myristate 13-acetate)
- Dibutyryl cAMP (Bucladesine) sodium
- Nedisertib (M3814)
- PLX5622
- IKE (Imidazole Ketone Erastin)
- STM2457
- Saruparib (AZD5305)
- New Products
- Contact Us
-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
-
research use only
AEE788 (NVP-AEE788) EGFR inhibitor
Cat.No.S1486
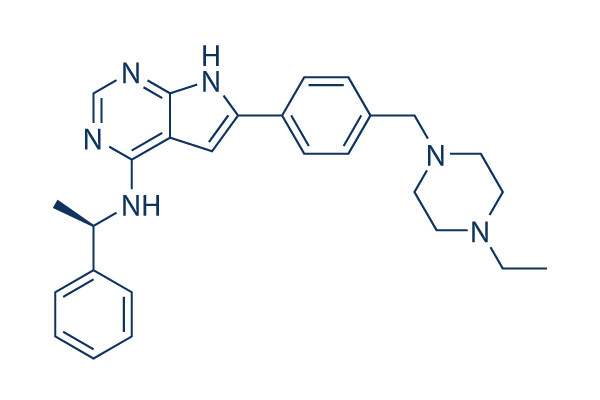
Chemical Structure
Molecular Weight: 440.58
Quality Control
Batch:
Purity:
99.62%
99.62
Cell Culture, Treatment & Working Concentration
| Cell Lines | Assay Type | Concentration | Incubation Time | Formulation | Activity Description | PMID |
|---|---|---|---|---|---|---|
| Huh7 | Cytotoxicity assay | Cytotoxicity against human hepatocellular carcinoma cell line (Huh7), CC50=27.83 Μm | ||||
| Jeko B cells | Function assay | 2 h | Inhibition of PI3Kdelta in human Jeko B cells assessed as inhibition of Akt phosphorylation after 2 hrs, IC50=0.017 μM | |||
| Click to View More Cell Line Experimental Data | ||||||
Chemical Information, Storage & Stability
| Molecular Weight | 440.58 | Formula | C27H32N6 |
Storage (From the date of receipt) | |
|---|---|---|---|---|---|
| CAS No. | 497839-62-0 | Download SDF | Storage of Stock Solutions |
|
|
| Synonyms | N/A | Smiles | CCN1CCN(CC1)CC2=CC=C(C=C2)C3=CC4=C(N3)N=CN=C4NC(C)C5=CC=CC=C5 | ||
Solubility
|
In vitro |
DMSO
: 88 mg/mL
(199.73 mM)
Water : Insoluble Ethanol : Insoluble |
Molarity Calculator
|
In vivo |
|||||
In vivo Formulation Calculator (Clear solution)
Step 1: Enter information below (Recommended: An additional animal making an allowance for loss during the experiment)
mg/kg
g
μL
Step 2: Enter the in vivo formulation (This is only the calculator, not formulation. Please contact us first if there is no in vivo formulation at the solubility Section.)
%
DMSO
%
%
Tween 80
%
ddH2O
%
DMSO
+
%
Calculation results:
Working concentration: mg/ml;
Method for preparing DMSO master liquid: mg drug pre-dissolved in μL DMSO ( Master liquid concentration mg/mL, Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug. )
Method for preparing in vivo formulation: Take μL DMSO master liquid, next addμL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O, mix and clarify.
Method for preparing in vivo formulation: Take μL DMSO master liquid, next add μL Corn oil, mix and clarify.
Note: 1. Please make sure the liquid is clear before adding the next solvent.
2. Be sure to add the solvent(s) in order. You must ensure that the solution obtained, in the previous addition, is a clear solution before proceeding to add the next solvent. Physical methods such
as vortex, ultrasound or hot water bath can be used to aid dissolving.
Mechanism of Action
| Targets/IC50/Ki | |
|---|---|
| In vitro |
AEE788 (NVP-AEE788) also inhibits KDR, c-abl, c-Src, and Flt-1 with IC50 of 50-80 nM. It is not sensitive to ErbB-4, PDGFR-β, Flt-3, Flt-4, RET, and c-Kit and has no inhibitory to Ins-R, IGF-1R, PKC-α, and PKA. This compound potently inhibits EGFR phosphorylation in A431 cells with IC50 of 11 nM. It also inhibits the phosphorylation of KDR in CHO cells and erbB2 in BT-474 cells, without any effects on PDGF-induced phosphorylation in A31 cells. It inhibits the proliferation of NCI-H596, MK, BT-474 and SK-BR-3 cells with IC50 of 78, 56, 49 and 381 nM, respectively. Otherwise, it has the additional property of inhibiting cellular proliferation driven by EGFR mutant including 32D/EGFR and 32D/EGFRvIII. It further also inhibits both EGF- and VEGF-driven HUVEC proliferation with IC50 of 43 and 155 nM, respectively. [1] It inhibits the phosphorylation of EGFR, VEGFR2, Akt, and MAPK in human cutaneous SCC cell lines (Colo16, HaCaT, SRB1, and SRB12 cells), which leads to growth inhibition and induction of apoptosis. [2] It inhibits the phosphorylation of EGFR and Akt in HT29 cells at 0.2 to 1.0 μM. [3] It inhibits cell proliferation and prevents EGF- and neuregulin-induced HER1, HER2, and HER3 activation in medulloblastoma cell lines. It shows growth-suppressive activities in chemosensitive and chemoresistant medulloblastoma cells. [4]
|
| Kinase Assay |
Protein Kinase Assays
|
|
The in vitro kinase assays for AEE788 (NVP-AEE788) are performed in 96-well plates (30 μL) at ambient temperature for 15–45 min using the recombinant glutathione S-transferase-fused kinase domains (4-100 ng, depending on specific activity). [γ33P]ATP is used as phosphate donor and polyGluTyr-(4:1) peptide as acceptor. With the exception of protein kinase C-α, cyclin-dependent kinase 1/cycB and protein kinase A are protamine sulfate (200 μg/mL), histone H1 (100 μg/mL), and the heptapeptide Leu-Arg-Arg-Ala-Ser-Leu-Gly (known as Kemptide Bachem) respectively and are used as peptide substrates. Assays are optimized for each kinase using the following ATP concentrations: 1.0 μM (c-Kit, c-Met, c-Fms, c-Raf-1, and RET), 2.0 μM (EGFR, erbB2, ErbB3, and ErbB4), 5.0 μM (c-abl), 8.0 μM (Flt-1, Flt-3, Flt-4, Flk, KDR, FGFR-1, and Tek), 10.0 μM (PDGFR-β, protein kinase C-α, and cyclin-dependent kinase 1), and 20.0 μM (c-Src and protein kinase A). The reaction is terminated by the addition of 20 μL 125 mM EDTA. Thirty μL (c-abl, c-Src, insulin-like growth factor-1R, RET-Men2A, and RET-Men2B) or 40 μL (all other kinases) of the reaction mixture is transferred onto Immobilon-polyvinylidene difluoride membrane, presoaked with 0.5% H3PO4 and mounted on a vacuum manifold. Vacuum is then applied and each well rinsed with 200 μL 0.5% H3PO4. Membranes are removed and washed four times with 1.0% H3PO4 and once with ethanol. Dried membranes are counted after mounting in a Packard TopCount 96-well frame and with the addition of 10 μL/well of Microscint. IC50 values (±SE) are calculated by linear regression analysis of the percentage inhibition and are averages of at least three determinations.
|
|
| In vivo |
AEE788 (NVP-AEE788) produces a dose-dependent inhibition of tumor growth in NCI-H596 or DU145 xenograft models, with only minor body weight changes. It induces tumor regression by 57% at 50 mg/kg in the NeuT/erbB2 GeMag model and potently inhibits EGF-induced EGFR phosphorylation in A431 tumors and erbB2 phosphorylation in GeMag tumors. This compound dose-dependently inhibited angiogenesis induced by VEGF and does not inhibit bFGF-induced angiogenesis. [1] It suppresses the growth of tumor volume by 54% in Colo16 xenografts at 50 mg/kg, which is due to the inhibition of phosphorylation of EGFR, VEGFR, Akt, and MAPK. [2] At 50 mg/kg, it also inhibits growth of tumors in the cecum and peritoneum (>50%) and reduces the incidence of lymph node metastasis to 70% in HT29 cells implanted in the cecum of nude mice, without loss of body weight and gross evidence of neovascularization. It significantly lowers the expression levels of pEGFR and pVEGFR in HT29 cecal tumors and does not alter those of EGF, VEGF, EGFR, or VEGFR. Combined with CPT-11, it results in significantly smaller tumors and complete inhibition of lymph node metastasis. [3] This compound inhibits the growth of Daoy, DaoyPt, and DaoyHER2 xenografts by 51%, 45%, and 72%, respectively. [4] It could promote LBH589-mediated generation of reactive oxygen species in K562 tumor cells, which in turn increase apoptosis. [5]
|
References |
|
Clinical Trial Information
(data from https://clinicaltrials.gov, updated on 2024-05-22)
| NCT Number | Recruitment | Conditions | Sponsor/Collaborators | Start Date | Phases |
|---|---|---|---|---|---|
| NCT00107237 | Completed | Brain and Central Nervous System Tumors |
Novartis Pharmaceuticals|Novartis |
October 2003 | Phase 1|Phase 2 |
| NCT00118456 | Completed | Cancer |
Novartis Pharmaceuticals|Novartis |
July 2003 | Phase 1 |
Tech Support
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
Signaling Pathway Map
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.