
- Bioactive Compounds
- By Signaling Pathways
- PI3K/Akt/mTOR
- Epigenetics
- Methylation
- Immunology & Inflammation
- Protein Tyrosine Kinase
- Angiogenesis
- Apoptosis
- Autophagy
- ER stress & UPR
- JAK/STAT
- MAPK
- Cytoskeletal Signaling
- Cell Cycle
- TGF-beta/Smad
- Compound Libraries
- Antibodies
- Bioreagents
- qPCR
- 2x SYBR Green qPCR Master Mix
- 2x SYBR Green qPCR Master Mix(Low ROX)
- 2x SYBR Green qPCR Master Mix(High ROX)
- Protein Assay
- Protein A/G Magnetic Beads for IP
- Anti-Flag magnetic beads
- Anti-Flag Affinity Gel
- Anti-Myc magnetic beads
- Anti-HA magnetic beads
- Poly FLAG Peptide lyophilized powder
- Protease Inhibitor Cocktail
- Protease Inhibitor Cocktail (EDTA-Free, 100X in DMSO)
- Phosphatase Inhibitor Cocktail (2 Tubes, 100X)
- Cell Biology
- Cell Counting Kit-8 (CCK-8)
- Animal Experiment
- Mouse Direct PCR Kit (For Genotyping)
- New Products
- Contact Us
-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
-
 Other Countries
Other Countries
PF-477736
Synonyms: PF-736, PF-00477736
PF-477736 (PF-736, PF-00477736) is a selective, potent and ATP-competitive Chk1 inhibitor with Ki of 0.49 nM in a cell-free assay and also inhibits VEGFR2, Aurora-A, FGFR3, Flt3, Fms (CSF1R), Ret and Yes. It shows ~100-fold selectivity for Chk1 than Chk2.
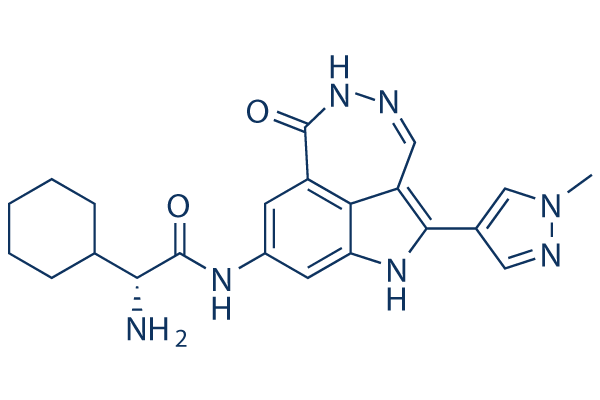
PF-477736 Chemical Structure
CAS: 952021-60-2
Selleck's PF-477736 has been cited by 29 Publications
1 Customer Review
Purity & Quality Control
Batch:
Purity:
99.80%
99.80
PF-477736 Related Products
| Related Targets | Chk1 Chk2 | Click to Expand |
|---|---|---|
| Related Compound Libraries | Kinase Inhibitor Library PI3K/Akt Inhibitor Library MAPK Inhibitor Library DNA Damage/DNA Repair compound Library Cell Cycle compound library | Click to Expand |
Signaling Pathway
Choose Selective Chk Inhibitors
Cell Data
| Cell Lines | Assay Type | Concentration | Incubation Time | Formulation | Activity Description | PMID |
|---|---|---|---|---|---|---|
| TC32 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for TC32 cells | 29435139 | |||
| U-2 OS | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for U-2 OS cells | 29435139 | |||
| A673 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for A673 cells | 29435139 | |||
| Saos-2 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for Saos-2 cells | 29435139 | |||
| RD | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for RD cells | 29435139 | |||
| SK-N-SH | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SK-N-SH cells | 29435139 | |||
| OHS-50 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for OHS-50 cells | 29435139 | |||
| SJ-GBM2 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SJ-GBM2 cells | 29435139 | |||
| SK-N-MC | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for SK-N-MC cells | 29435139 | |||
| NB-EBc1 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for NB-EBc1 cells | 29435139 | |||
| LAN-5 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for LAN-5 cells | 29435139 | |||
| Click to View More Cell Line Experimental Data | ||||||
Biological Activity
| Description | PF-477736 (PF-736, PF-00477736) is a selective, potent and ATP-competitive Chk1 inhibitor with Ki of 0.49 nM in a cell-free assay and also inhibits VEGFR2, Aurora-A, FGFR3, Flt3, Fms (CSF1R), Ret and Yes. It shows ~100-fold selectivity for Chk1 than Chk2. | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Targets |
|
| In vitro | ||||
| In vitro | PF-477736 (128 nM) abrogates the camptothecin-induced DNA damage checkpoint in a dose-dependent manner in CA46 and HeLa cells. PF-477736 effectively abrogates the gemcitabine-induced S-phase arrest with a corresponding increase in apoptotic cell populations in HT29 cells. PF-477736 (540 nM) enhances gemcitabine-induced cytotoxicity in a time- and dose-dependent manner in HT29 cells. PF-477736 potentiates the growth-inhibitory activity of a panel of chemotherapeutic agents across a broad spectrum of p53-deficient human cancer cell lines in the MTT assay. Addition of PF-477736 (360 nM) to gemcitabine-arrested cells induces a dramatic increase in the intensity of H2AX phosphorylation, reflecting a greater number of γ-H2AX molecules near sites of DNA damage. [1] PF-477736 (0.5 nM) selectively blocks p73 and P53 phosphorylation in presence of curcumin in HL-60 cells. [2] PF-477736 (360 nM) suppresses docetaxel-induced phosphorylation of histone H3 (Ser10) and Cdc25C (Ser216) and potentiates apoptosis in COLO205 cells. [3] PF-477736 (250 nM) combined with MK-1775 has marked synergistic cytotoxic activity in OVCAR-5 cells. PF-477736 (250 nM) combined with MK-1775 causes accumulation of cells with a DNA content between 2N and 4N in OVCAR-5 cells. PF-477736 (250 nM) combined with MK-1775 causes premature mitosis before the end of DNA replication, with damaged DNA leading to apoptotic cell death in OVCAR-5 cells. [4] |
|||
|---|---|---|---|---|
| Kinase Assay | Binding assay | |||
| The assay is performed in a 96-well plate for 20 minutes at 30℃ in 0.1 mL of assay buffer containing 50 mM TRIS pH 7.5, 0.4 M NaCl, 4 mM PEP, 0.15 mM NADH, 28 units of lactate dehydrogenase/mL, 16 units of pyruvate kinase/mL, 3 mM DTT, 0.125 mM Syntide-2, 0.15 mM ATP and 25 mM magnesium chloride. Assays are initiated with 1 nM of CHK1 kinase domain. The inhibition of CHK1 activity is determined by measuring initial velocities in the presence of varying concentrations of PF-477736. The data is analyzed using Enzyme Kinetic and Excel software and fit to a kinetic model for competitive inhibition to obtain a Ki value. The kinase selectivity of PF-477736 is evaluated by screening the compound at 1 μM or 10 μM against a panel 2 of about 100 protein kinases. | ||||
| Cell Research | Cell lines | HT29, Colo205, PC-3, MDA-MB-231 and K562 cells | ||
| Concentrations | ~1 μM | |||
| Incubation Time | 96 hours | |||
| Method | The IC50 assay measures the antiproliferative effects of PF-477736 on p53-defective human cancer cell lines. Cells in each line are seeded in complete medium at an exponentially growing density in 96-well assay plate and allowed to attach for 16 hours. Serial dilutions of PF-477736 are then done, and appropriate controls are added to each plate. Cells are incubated with drug for 96 hours. After incubation, MTT working stock diluted in complete medium is added to each well, and cells are incubated for 4 hours. After centrifugation and supernatant removal, DMSO is added to each well and plates are read on SpectraMax plate reader at 540 nm. |
|||
| Experimental Result Images | Methods | Biomarkers | Images | PMID |
| Western blot | p-53 / p-p53 / p21 / MDM2 p-ATM / γ-H2AX |
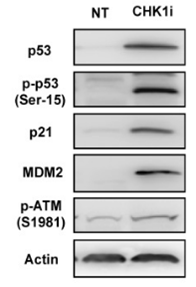
|
31362335 | |
| Growth inhibition assay | Cell viability |
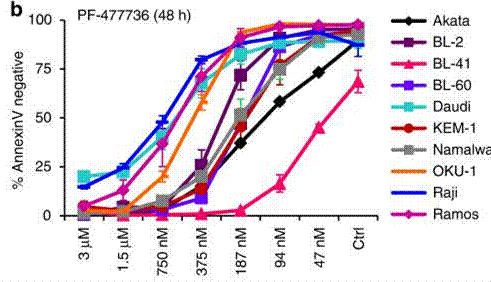
|
29167438 | |
| Immunofluorescence | p-p53 |
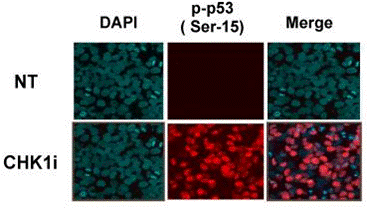
|
31362335 | |
| In Vivo | ||
| In vivo |
PF-477736 (4 mg/kg i.v.) results in terminal half-life (T1/2) of 2.9 hours, AUC of 5.72 μg×hr/mL and CLp of 11.8 mL/min/kg in rats. PF-477736 dose-dependently enhances the antitumor activity of a maximum tolerated dose of gemcitabine in the Colo205 xenograft mouse model. PF-477736 (12 mg/kg) induces an increase in the phosphorylation of histone H3 (Ser10) and of phospho-histone H2AX in the Colo205 xenograft mouse model. [1] PF-477736 (15 mg/kg i.p.) enhances docetaxel induced tumor growth inhibition and tumor growth delay in COLO205 and MDA-MB-231 xenograft models. [3] PF 477736 (10 mg/kg once daily i.p.) combined with MK-1775 (30 mg/kg twice a day oral) leads to greater tumor growth inhibition in mice bearing OVCAR-5 xenografts. [4] |
|
|---|---|---|
| Animal Research | Animal Models | Colo205 xenograft mouse model |
| Dosages | 40 mg/kg | |
| Administration | intravenous injection | |
| NCT Number | Recruitment | Conditions | Sponsor/Collaborators | Start Date | Phases |
|---|---|---|---|---|---|
| NCT00437203 | Terminated | Neoplasms |
Pfizer |
December 2006 | Phase 1 |
Chemical Information & Solubility
| Molecular Weight | 419.48 | Formula | C22H25N7O2 |
| CAS No. | 952021-60-2 | SDF | Download PF-477736 SDF |
| Smiles | CN1C=C(C=N1)C2=C3C=NNC(=O)C4=C3C(=CC(=C4)NC(=O)C(C5CCCCC5)N)N2 | ||
| Storage (From the date of receipt) | |||
|
In vitro |
DMSO : 84 mg/mL ( (200.24 mM); Moisture-absorbing DMSO reduces solubility. Please use fresh DMSO.) Water : Insoluble Ethanol : Insoluble |
Molecular Weight Calculator |
|
In vivo Add solvents to the product individually and in order. |
In vivo Formulation Calculator |
||||
Preparing Stock Solutions
Molarity Calculator
In vivo Formulation Calculator (Clear solution)
Step 1: Enter information below (Recommended: An additional animal making an allowance for loss during the experiment)
mg/kg
g
μL
Step 2: Enter the in vivo formulation (This is only the calculator, not formulation. Please contact us first if there is no in vivo formulation at the solubility Section.)
% DMSO
%
% Tween 80
% ddH2O
%DMSO
%
Calculation results:
Working concentration: mg/ml;
Method for preparing DMSO master liquid: mg drug pre-dissolved in μL DMSO ( Master liquid concentration mg/mL, Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug. )
Method for preparing in vivo formulation: Take μL DMSO master liquid, next addμL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O, mix and clarify.
Method for preparing in vivo formulation: Take μL DMSO master liquid, next add μL Corn oil, mix and clarify.
Note: 1. Please make sure the liquid is clear before adding the next solvent.
2. Be sure to add the solvent(s) in order. You must ensure that the solution obtained, in the previous addition, is a clear solution before proceeding to add the next solvent. Physical methods such
as vortex, ultrasound or hot water bath can be used to aid dissolving.
Tech Support
Answers to questions you may have can be found in the inhibitor handling instructions. Topics include how to prepare stock solutions, how to store inhibitors, and issues that need special attention for cell-based assays and animal experiments.
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
* Indicates a Required Field
Frequently Asked Questions
Question 1:
Can you advise the vehicle suggested for S2904: 2% DMSO/40% PEG 300 at 5mg/ml, is it a suspension or clear solution? I wanted to know how to dissolve this compound for an i.p. injection in mice
Answer:
S2904 can dissolve in 2% DMSO/40% PEG 300 at 5mg/ml as a clear solution. If you are going to use this kind of vehicle, please dissolve the compound in DMSO clearly first, then add PEG300, then water.
Tags: buy PF-477736 | PF-477736 supplier | purchase PF-477736 | PF-477736 cost | PF-477736 manufacturer | order PF-477736 | PF-477736 distributor
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.