-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
research use only
Canagliflozin (JNJ-28431754) SGLT2 Inhibitor
Cat.No.S2760
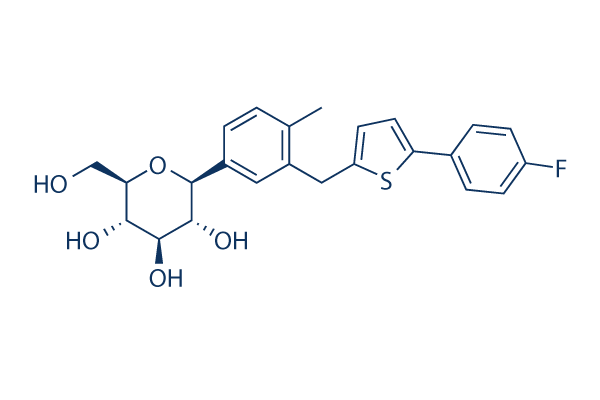
Chemical Structure
Molecular Weight: 444.52
Quality Control
Cell Culture, Treatment & Working Concentration
| Cell Lines | Assay Type | Concentration | Incubation Time | Formulation | Activity Description | PMID |
|---|---|---|---|---|---|---|
| CHO | Function assay | 120 mins | Inhibition of human SGLT2 expressed in CHO cells assessed as decrease in uptake of [14C]AMG after 120 mins by TopCount method, IC50 = 0.0022 μM. | 28447791 | ||
| CHO | Function assay | 120 mins | Inhibition of human SGLT1 expressed in CHO cells assessed as decrease in uptake of [14C]AMG after 120 mins by TopCount method, IC50 = 0.265 μM. | 28447791 | ||
| CHO-K1 | Function assay | Inhibition of human SGLT2 expressed in CHO-K1 cells by [14C]AMG uptake assay, IC50 = 0.0067 μM. | 22652255 | |||
| CHO-K1 | Function assay | Inhibition of human SGLT1 expressed in CHO-K1 cells by [14C]AMG uptake assay, IC50 = 1.9 μM. | 22652255 | |||
| Click to View More Cell Line Experimental Data | ||||||
Solubility
|
In vitro |
DMSO
: 89 mg/mL
(200.21 mM)
Water : Insoluble Ethanol : Insoluble |
Molarity Calculator
|
In vivo |
|||||
In vivo Formulation Calculator (Clear solution)
Step 1: Enter information below (Recommended: An additional animal making an allowance for loss during the experiment)
Step 2: Enter the in vivo formulation (This is only the calculator, not formulation. Please contact us first if there is no in vivo formulation at the solubility Section.)
Calculation results:
Working concentration: mg/ml;
Method for preparing DMSO master liquid: mg drug pre-dissolved in μL DMSO ( Master liquid concentration mg/mL, Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug. )
Method for preparing in vivo formulation: Take μL DMSO master liquid, next addμL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O, mix and clarify.
Method for preparing in vivo formulation: Take μL DMSO master liquid, next add μL Corn oil, mix and clarify.
Note: 1. Please make sure the liquid is clear before adding the next solvent.
2. Be sure to add the solvent(s) in order. You must ensure that the solution obtained, in the previous addition, is a clear solution before proceeding to add the next solvent. Physical methods such
as vortex, ultrasound or hot water bath can be used to aid dissolving.
Chemical Information, Storage & Stability
| Molecular Weight | 444.52 | Formula | C24H25FO5S |
Storage (From the date of receipt) | |
|---|---|---|---|---|---|
| CAS No. | 842133-18-0 | Download SDF | Storage of Stock Solutions |
|
|
| Synonyms | TA 7284 | Smiles | CC1=C(C=C(C=C1)C2C(C(C(C(O2)CO)O)O)O)CC3=CC=C(S3)C4=CC=C(C=C4)F | ||
Mechanism of Action
| Targets/IC50/Ki |
mSGLT2
(Cell-free assay) 2 nM
rSGLT2
(Cell-free assay) 3.7 nM
hSGLT2
(Cell-free assay) 4.4 nM
|
|---|---|
| In vitro |
Canagliflozin (JNJ 28431754) is a novel C-glucoside with thiophene ring. It inhibits Na+-dependent 14C-AMG uptake in a concentration-dependent fashion. This compound inhibits 14C-AMG uptake in CHO-hSGLT1 and mSGLT1 cells with IC50 of 0.7 μM and >1 μM, respectively. It inhibits the facilitative (non-Na+-linked) GLUT-mediated 2H-2-DG uptake in L6 myoblasts by less than 50%. In sham-injected oocytes, Canagliflozin (10 μM) or phlorizin (3 mM) alone in the presence of 50 μM DNJ does not affect currents. In SGLT3-injected oocytes, DMSO and Canagliflozin 10 μM inhibits DNJ-induced currents by 15.6% and 23.4%, respectively.
|
| In vivo |
Canagliflozin (JNJ 28431754) shows pronounced anti-hyperglycemic effects in high-fat diet fed KK (HF-KK) mice. Oral administration at 30 mg/kg of this compound to male SD rats induces glucose excretion over 24 hours by 3,696 mg per 200 g body weight. Pharmacokinetic studies reveals a much higher exposure following oral administration. Following intravenous and oral doses of 3 and 10 mg/kg, respectively, to male SD rats, AUC0−inf, po, t1/2 and oral bioavailability are determined to be 35,980 ng·h/mL, 5.2 hours, and 85%, respectively. Thus, inhibition of SGLT2 in renal tubules after oral dosing is likely to continuously suppress reabsorption of glucose. The extensive UGE would reflect excellent pharmacokinetic properties in vivo as well as high potency of SGLT2 inhibition. Since most of the filtered glucose is reabsorbed by SGLT2 in the renal tubules, the novel compound would be useful for an anti-diabetic agent. Single oral administration at 3 mg/kg remarkably reduced blood glucose levels without influencing food intake in hyperglycemic high-fat diet fed KK (HF-KK) mice. There is a 48% reduction in blood glucose level versus vehicle at 6 hours. In contrast, it only slightly affects blood glucose levels in normoglycemic mice. Therefore, Canagliflozin would control hyperglycemia in the therapy of T2DM with low risk of hypoglycemia.
|
References |
|
Applications
| Methods | Biomarkers | Images | PMID |
|---|---|---|---|
| Western blot | p-β-catenin / β-catenin / Cyclin D1 pACC / ACC / p-AMPKα / AMPKα / p-S6K / S6K / p-S6 / S6 |
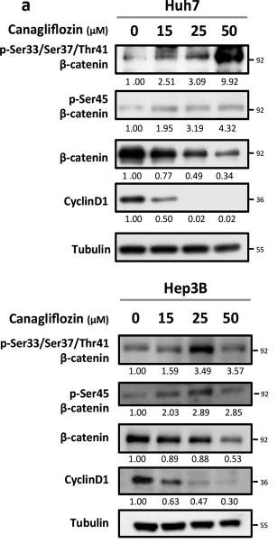
|
31142735 |
Clinical Trial Information
(data from https://clinicaltrials.gov, updated on 2024-05-22)
| NCT Number | Recruitment | Conditions | Sponsor/Collaborators | Start Date | Phases |
|---|---|---|---|---|---|
| NCT06301529 | Enrolling by invitation | Healthy Aging |
AgelessRx |
February 12 2024 | Phase 4 |
| NCT04720859 | Unknown status | Postprandial Hypoglycemia |
Hospital Universitari Vall d''Hebron Research Institute |
January 5 2018 | Not Applicable |
| NCT02891954 | Enrolling by invitation | Diabetes Mellitus Type 2 |
University of Maryland Baltimore|National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) |
September 2016 | Phase 1 |
| NCT02737657 | Completed | Diabetes Mellitus Type 2 |
Janssen-Cilag International NV |
April 2016 | -- |
| NCT02857764 | Completed | Diabetes Mellitus Type 2 |
Janssen Research & Development LLC |
February 15 2016 | -- |
Tech Support
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
Signaling Pathway Map
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.