-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
research use only
Complex I Immunocapture Antibody [K15L2]
Cat.No.: F2553
Application:
Reactivity:
-
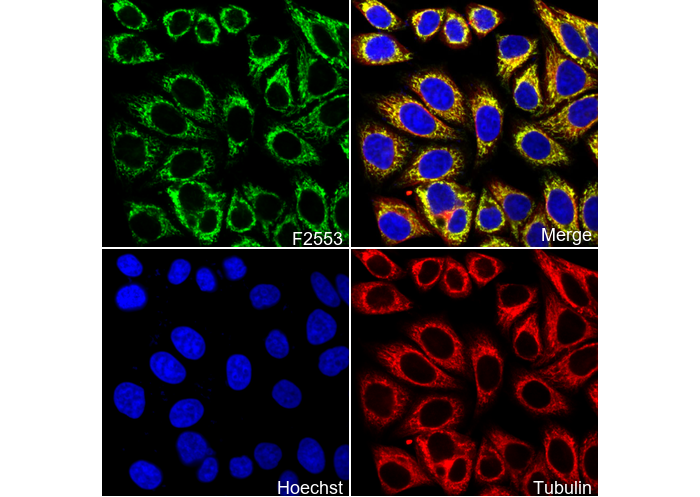 Immunofluorescent analysis of HepG2 cells using F2553 (green, 1:1000), Hoechst (blue) and tubulin (Red).
Immunofluorescent analysis of HepG2 cells using F2553 (green, 1:1000), Hoechst (blue) and tubulin (Red).
Usage Information
| Dilution |
|---|
|
| Application |
|---|
| IP, IF, FCM |
| Reactivity |
|---|
| Mouse, Rat, Cow, Human |
| Source |
|---|
| Mouse Monoclonal Antibody |
| Storage Buffer |
|---|
| PBS, pH 7.2+50% Glycerol+0.05% BSA+0.01% NaN3 |
| Storage (from the date of receipt) |
|---|
| -20°C (avoid freeze-thaw cycles), 2 years |
| Positive Control | Human heart mitochondria; Bovine heart mitochondria; Mouse heart mitochondria; Mouse brain mitochondria; Fibroblasts cells; HL-60 cells; HepG2 cells |
|---|---|
| Negative Control |
Experimental Methods
| IF |
|---|
Experimental Protocol:
Sample Preparation
1. Adherent Cells: Place a clean, sterile coverslip in a culture dish. Once the cells grow to near confluence as a monolayer, remove the coverslip for further use.
2. Suspension Cells: Seed the cells onto a clean, sterile slide coated with poly-L-lysine.
3. Frozen Sections: Allow the slide to thaw at room temperature. Wash it with pure water or PBS for 2 times, 3 minutes each time.
4. Paraffin Sections: Deparaffinization and rehydration. Wash the slide with pure water or PBS for 3 times, 3 minutes each time. Then perform antigen retrieval.
Fixation
1. Fix the cell coverslips/spots or tissue sections at room temperature using a fixative such as 4% paraformaldehyde (4% PFA) for 10-15 minutes.
2. Wash the sample with PBS for 3 times, 3 minutes each time.
Permeabilization
1.Add a detergent such as 0.1–0.3% Triton X-100 to the sample and incubate at room temperature for 10–20 minutes.
(Note: This step is only required for intracellular antigens. For antigens expressed on the cell membrane, this step is unnecessary.)
Wash the sample with PBS for 3 times, 3 minutes each time.
Blocking
Add blocking solution and incubate at room temperature for at least 1 hour. (Common blocking solutions include: serum from the same source as the secondary antibody, BSA, or goat serum.)
Note: Ensure the sample remains moist during and after the blocking step to prevent drying, which can lead to high background.
Immunofluorescence Staining (Day 1)
1. Remove the blocking solution and add the diluted primary antibody.
2. Incubate the sample in a humidified chamber at 4°C overnight.
Immunofluorescence Staining (Day 2)
1. Remove the primary antibody and wash with PBST for 3 times, 5 minutes each time.
2. Add the diluted fluorescent secondary antibody and incubate in the dark at 4°C for 1–2 hours.
3. Remove the secondary antibody and wash with PBST for 3 times, 5 minutes each time.
4. Add diluted DAPI and incubate at room temperature in the dark for 5–10 minutes.
5. Wash with PBST for 3 times, 5 minutes each time.
Mounting
1. Mount the sample with an anti-fade mounting medium.
2. Allow the slide to dry at room temperature overnight in the dark.
3. Store the slide in a slide storage box at 4°C, protected from light.
|
Biological Description
| Specificity |
|---|
| Complex I Immunocapture Antibody [K15L2] detects endogenous levels of total Complex I Immunocapture protein. |
| Clone |
|---|
| K15L2 |
| Background |
|---|
| Complex I (NADH:ubiquinone oxidoreductase) is the largest, L-shaped enzyme complex in the mitochondrial inner membrane, comprising about 45 subunits, with 14 core subunits conserved across species. Its hydrophilic peripheral arm contains the FMN cofactor and a relay of 8–9 iron-sulfur (FeS) clusters (such as N3, N1b, N4, N5, N6a/b, N2) for electron transfer, while the membrane arm features four proton-pumping modules (P_D, P_P, P_NuoL, P_NuoM) powered by charged residues and helical rearrangements. Electrons from matrix NADH enter at the N-module (NDUFV1/2), traverse ~90 Å along the FeS clusters to the Q-module’s N2 [4Fe-4S] cluster near NDUFS2/7, and reduce ubiquinone in a ~30 Å tunnel via semiquinone intermediates. The redox reaction (~-150 mV driving force) is tightly coupled to vectorial translocation of four protons per two electrons through conformational waves, ubiquinone binding elicits a two-state stabilization, rigidifying the complex and propagating allosteric shifts from the Q-site to distal pumps via antiporter-like helices (TMH 38–42 in ND2/4/5), thereby generating membrane potential (Δψ) for ATP synthesis and minimizing superoxide production by keeping the N2-Q distance short (~7 Å). As the entry point for oxidative phosphorylation, Complex I maintains cellular energy balance, links the TCA cycle and NADH homeostasis, and influences ROS signaling and mitochondrial DNA maintenance. Dysfunction due to mutations (e.g., NDUFS4, ND1) or assembly defects leads to elevated ROS, impaired bioenergetics, and diseases such as Leigh syndrome, MELAS, Parkinson’s disease (via α-synuclein inhibition), and Leber’s hereditary optic neuropathy, particularly impacting neurons with high energy demands. |
| References |
|---|
|
Tech Support
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.