TGF-beta/Smad inhibitors are a class of molecular agents designed to modulate the transforming growth factor-beta (TGF-β)/Smad signaling cascade, one of the most evolutionarily conserved and functionally pleiotropic pathways in eukaryotic cells. Over the past three decades, research into these inhibitors has exploded, driven by the pathway’s dual role in tissue homeostasis and disease pathogenesis—acting as a tumor suppressor in early-stage cancer while promoting metastasis and drug resistance in advanced malignancies, and mediating excessive extracellular matrix (ECM) deposition in fibrotic disorders. This review focuses on the scientific underpinnings of TGF-beta/Smad inhibitors, their molecular targeting strategies, preclinical efficacy in disease models, and emerging challenges in translational research.
-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
TGF-beta/Smad Inhibitors/Activators
Other TGF-beta/Smad Related Inhibitors
Isoform-selective Products
| Cat.No. | Product Name | Information | Product Use Citations | Product Validations |
|---|---|---|---|---|
| S1067 | SB431542 | SB431542 is a potent and selective inhibitor of ALK5 with IC50 of 94 nM in a cell-free assay, 100-fold more selective for ALK5 than p38 MAPK and other kinases. |
|
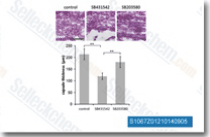
|
| S7223 | RepSox (E-616452) | RepSox (E-616452, SJN 2511, ALK5 Inhibitor II) is a potent and selective inhibitor of the TGFβR-1/ALK5 with IC50 of 23 nM and 4 nM for ATP binding to ALK5 and ALK5 autophosphorylation in cell-free assays, respectively. |
|

|
| S7530 | Vactosertib (TEW-7197) | Vactosertib (TEW-7197, EW-7197) is a highly potent, selective, and orally bioavailable TGF-β receptor ALK4/ALK5 inhibitor with IC50 of 13 nM and 11 nM, respectively. Phase 1. |
|

|
| S2618 | LDN-193189 | LDN-193189 (DM3189) is a selective BMP signaling inhibitor, inhibits the ALK1, ALK2, ALK3 and ALK6 with IC50s of 0.8 nM, 0.8 nM, 5.3 nM and 16.7 nM in the kinase assay, respectively. LDN-193189 inhibits the transcriptional activity of the BMP type I receptors ALK2 and ALK3 with IC50s of 5 nM and 30 nM in C2C12 cells, respectively, exhibits 200-fold selectivity for BMP versus TGF-β. For cell testing, the water-soluble S7507 LDN-193189 2HCl is recommended.This product has poor solubility, animal experiments are available, cell experiments please choose carefully! |
|

|
| S7692 | A-83-01 | A-83-01 is a potent inhibitor of TGF-β type I receptor (ALK5-TD) with IC50 of 12 nM. This compound also inhibits the transcription induced by activin/nodal type I receptor (ALK4-TD) and nodal type I receptor (ALK7-TD) with IC50 of 45 nM and 7.5 nM, respectively.Solutions are unstable and should be fresh-prepared. |
|
|
| S7507 | LDN-193189 Dihydrochloride | LDN-193189 (DM3189) 2HCl is a selective BMP signaling inhibitor, inhibits the ALK1, ALK2, ALK3 and ALK6 with IC50s of 0.8 nM, 0.8 nM, 5.3 nM and 16.7 nM in the kinase assay, respectively. LDN-193189 inhibits the transcriptional activity of the BMP type I receptors ALK2 and ALK3 with IC50s of 5 nM and 30 nM in C2C12 cells, respectively, exhibits 200-fold selectivity for BMP versus TGF-β. |
|
|
| S2230 | Galunisertib (LY2157299) | Galunisertib (LY2157299) is a potent TGFβ receptor I (TβRI / ALK5) inhibitor with IC50 of 56 nM in a cell-free assay. Phase 2/3. |
|

|
| S6654 | SRI-011381 (C381) | SRI-011381 (C381), a novel agonist of the TGF-beta signaling pathway for treatment of Alzheimer's disease, physically targets the lysosome, promotes lysosomal acidification, increases breakdown of lysosomal cargo, and improves lysosome resilience to damage. |
|
|
| S2704 | LY2109761 | LY2109761 is a novel selective TGF-β receptor type I/II (TβRI/II) dual inhibitor with Ki of 38 nM and 300 nM in a cell-free assay, respectively; shown to negatively affect the phosphorylation of Smad2. This compound blocks autophagy and induces apoptosis. |
|

|
| S7959 | SIS3 Hydrochloride | SIS3, a novel specific inhibitor of Smad3, inhibits TGF-β and activin signaling by suppressing Smad3 phosphorylation without affecting the MAPK/p38, ERK, or PI3-kinase signaling pathways. |
|
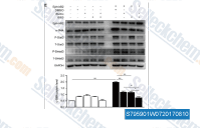
|
Signaling Pathway Map
The TGF-β/Smad signaling pathway: Core mechanisms and regulation
To contextualize the design and function of TGF-beta/Smad inhibitors, a foundational understanding of the TGF-β/Smad signaling pathway is essential. The pathway is activated by the binding of TGF-β ligands (TGF-β1, TGF-β2, TGF-β3) to transmembrane serine/threonine kinase receptors (TGFBR1/ALK5 and TGFBR2), which form a heterotetrameric complex upon ligand engagement.
Smad protein-mediated canonical signaling
Upon receptor activation, the intracellular Smad proteins—classified as receptor-regulated Smads (R-Smads: Smad2, Smad3), common-mediator Smad (Co-Smad: Smad4), and inhibitory Smads (I-Smads: Smad6, Smad7)—orchestrate the canonical signaling cascade. Phosphorylation of Smad2/3 by TGFBR1 triggers their dissociation from the receptor and heterodimerization with Smad4. This Smad complex translocates to the nucleus, where it interacts with transcription factors, co-activators, or co-repressors to regulate the expression of target genes involved in cell proliferation, differentiation, and apoptosis. I-Smads (Smad6/7) act as negative regulators by binding to activated TGFBR1, preventing R-Smad phosphorylation, or promoting ubiquitination and degradation of receptor complexes.
Non-canonical crosstalk and pathway plasticity
Beyond Smad-dependent signaling, TGF-β engages non-canonical pathways (e.g., MAPK, PI3K/Akt, Wnt / β-catenin) that modulate Smad function and expand the pathway’s biological output. This crosstalk is cell-type and context-dependent, and dysregulation of these interactions is a hallmark of pathological states. For example, in epithelial cells, TGF-β-induced Smad3 activation represses MYC expression to inhibit proliferation, while in mesenchymal cells, Smad3 cooperates with NF-κB to promote pro-fibrotic gene expression. Understanding this plasticity is critical for developing TGF-beta/Smad inhibitors that target disease-specific signaling nodes without disrupting physiological homeostasis.
Function of TGF-beta/Smad inhibitors in disease models
TGF-beta/Smad inhibitors are engineered to disrupt aberrant TGF-β/Smad signaling, with preclinical research focusing on two major disease areas: cancer and fibrosis. These inhibitors act through diverse mechanisms, including receptor kinase inhibition, Smad protein sequestration, and antisense targeting of TGF-β or Smad gene transcripts.
Anti-cancer activity of TGF-beta/Smad inhibitors
The dual role of TGF-β in cancer creates a paradox for therapeutic targeting: while early-stage tumors benefit from TGF-β’s tumor-suppressive effects, advanced metastatic disease is driven by TGF-β-mediated epithelial-mesenchymal transition (EMT), immune suppression, and angiogenesis. TGF-beta/Smad inhibitors address this by selectively blocking the pro-tumorigenic arm of the pathway. Preclinical studies have demonstrated that small-molecule ALK5 inhibitors (e.g., galunisertib, SB431542) suppress Smad2/3 phosphorylation in breast, lung, and pancreatic cancer models, reversing EMT and reducing metastatic burden. Additionally, Smad4-targeted inhibitors disrupt the nuclear translocation of Smad complexes, inhibiting the expression of pro-metastatic genes such as SNAI1 and TWIST1. Combination therapies—pairing TGF-beta/Smad inhibitors with immune checkpoint blockers (e.g., anti-PD-1)—have shown synergistic effects by reversing TGF-β-mediated immune evasion in the tumor microenvironment, restoring cytotoxic T-cell infiltration and activity. However, challenges remain, including dose-limiting toxicities (e.g., gastrointestinal perforation) due to off-target inhibition of physiological TGF-β signaling in normal tissues.
Anti-fibrotic effects of TGF-beta/Smad inhibitors
Fibrosis is characterized by excessive ECM deposition, driven by persistent TGF-β/Smad signaling in fibroblasts and myofibroblasts. TGF-beta/Smad inhibitors target this by blocking Smad3-mediated transcription of pro-fibrotic genes (e.g., COL1A1, CTGF, TIMP1) that encode collagens and ECM remodeling enzymes. In preclinical models of lung, liver, and renal fibrosis, ALK5 inhibitors reduce Smad3 phosphorylation and collagen accumulation, improving organ function and reducing scar tissue formation. Antisense oligonucleotides targeting TGF-β1 mRNA or Smad3 gene expression have shown specificity and reduced off-target effects compared to small-molecule inhibitors, as they directly silence the expression of key pathway components. For example, a Smad3 antisense oligonucleotide (SIS3) has been shown to prevent renal fibrosis in diabetic nephropathy models by inhibiting Smad3 binding to the COL1A1 promoter, without affecting Smad2 or Smad4 function. However, long-term efficacy is limited by the adaptive upregulation of alternative pro-fibrotic pathways (e.g., YAP /TAZ), highlighting the need for combinatorial inhibition of Smad-dependent and independent signaling.
Gene-targeted TGF-beta/Smad inhibitors: Next-generation strategies
Advancements in gene editing and nucleic acid therapeutics have enabled the development of precision TGF-beta/Smad inhibitors that target the genetic drivers of pathway dysregulation. These approaches offer improved specificity compared to small-molecule inhibitors, reducing off-target effects and enhancing therapeutic windows.
CRISPR/Cas9-mediated knockout of Smad and TGF-β pathway genes
CRISPR/Cas9 technology allows for the targeted knockout of Smad genes (e.g., SMAD3, SMAD4) or TGF-β receptor genes (TGFBR1, TGFBR2) in diseased cells, providing a tool to dissect pathway function and validate therapeutic targets. In preclinical cancer models, CRISPR-mediated SMAD4 knockout in pancreatic cancer cells abrogates Smad complex formation, inhibiting tumor growth and sensitizing cells to chemotherapy. Similarly, SMAD3 knockout in fibrotic liver models reduces pro-fibrotic gene expression and ECM deposition, reversing liver cirrhosis. While gene editing is currently limited to ex vivo applications (e.g., autologous cell therapy), in vivo delivery systems (e.g., lipid nanoparticles, viral vectors) are being optimized to enable systemic targeting of TGF-beta/Smad pathway genes.
RNA-based inhibitors of Smad gene expression
Small interfering RNAs (siRNAs) and microRNAs (miRNAs) targeting Smad or TGF-β gene transcripts represent a promising class of TGF-beta/Smad inhibitors. siRNAs against SMAD2/3 or TGFB1 have been shown to silence target gene expression in vitro and in vivo, reducing Smad phosphorylation and pro-fibrotic/cancer-associated gene expression. For example, lipid nanoparticle-delivered siRNA targeting TGFB1 has demonstrated efficacy in murine lung fibrosis models, with sustained reduction in TGF-β1 protein levels and collagen deposition for up to 4 weeks post-administration. miRNAs such as miR-29, which represses COL1A1 and SMAD3 expression, are being explored as endogenous TGF-beta/Smad inhibitors; replacement of miR-29 in fibrotic tissues restores its anti-fibrotic function, complementing exogenous inhibitor therapies.
Translational challenges and future directions
Despite robust preclinical data, translating TGF-beta/Smad inhibitors to clinical practice faces significant hurdles. First, the context-dependent function of TGF-β requires patient stratification—biomarkers such as Smad4 expression, TGF-β ligand levels, or EMT status are needed to identify patients most likely to benefit from inhibition. Second, systemic inhibition of TGF-β signaling can lead to adverse effects (e.g., impaired wound healing, increased infection risk) due to the pathway’s role in immune regulation and tissue repair. Tissue-specific delivery systems (e.g., antibody-drug conjugates, nanoparticle targeting) are being developed to restrict inhibitor activity to diseased tissues. Third, resistance to TGF-beta/Smad inhibitors is emerging as a challenge, driven by adaptive upregulation of alternative signaling pathways or mutations in Smad genes (e.g., SMAD4 loss in colorectal cancer). Combinatorial strategies targeting both Smad-dependent and independent pathways are being tested to overcome resistance.Looking forward, single-cell RNA sequencing and spatial transcriptomics are enabling the identification of cell-type-specific Smad gene expression patterns, facilitating the design of inhibitors tailored to distinct disease microenvironments. Additionally, personalized medicine approaches—using patient-derived organoids to test TGF-beta/Smad inhibitor efficacy—are improving the predictability of clinical outcomes. As our understanding of TGF-β/Smad signaling complexity deepens, TGF-beta/Smad inhibitors are poised to move from preclinical research to clinical application, offering new hope for patients with cancer and fibrotic disorders.
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.