-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
Chk Inhibitors
| Cat.No. | Product Name | Information | Product Use Citations | Product Validations |
|---|---|---|---|---|
| S1532 | AZD7762 | AZD7762 is a potent and selective inhibitor of Chk1 with IC50 of 5 nM in a cell-free assay. It is equally potent against Chk2 and less potent against CAM, Yes, Fyn, Lyn, Hck and Lck. Phase 1. |
|

|
| S2626 | Rabusertib (LY2603618) | Rabusertib (LY2603618, IC-83) is a highly selective Chk1 inhibitor with potential anti-tumor activity in a cell-free assay. IC50=7 nM, showing approximately 100-fold more potent against Chk1 than against any of the other protein kinases evaluated. Rabusertib (LY2603618) induces cell cycle arrest, DNA damage response and autophagy in cancer cells. Rabusertib (LY2603618) induces bak-dependent apoptosis in AML cell lines. |
|
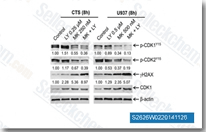
|
| S2735 | MK-8776 (SCH 900776) | MK-8776 (SCH 900776) is a selective Chk1 inhibitor with IC50 of 3 nM in a cell-free assay, showing 500-fold selectivity against Chk2. It is in Phase 2. |
|
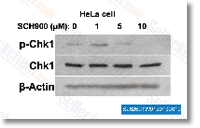
|
| S2683 | CHIR-124 | CHIR-124 is a novel and potent Chk1 inhibitor with IC50 of 0.3 nM in a cell-free assay. It shows 2,000-fold selectivity against Chk2, 500- to 5,000-fold less activity against CDK2/4 and Cdc2. |
|
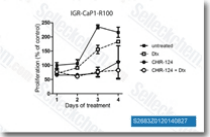
|
| S7178 | Prexasertib (LY2606368) Dihydrochloride | Prexasertib HCl (LY2606368) is an ATP-competitive CHK1 inhibitor with a Ki value of 0.9 nmol/L. For CHK2 and RSK, its IC50 values are 8 nM and 9 nM respectively in cell-free assay. |
|

|
| S2904 | PF-477736 | PF-477736 (PF-736, PF-00477736) is a selective, potent and ATP-competitive Chk1 inhibitor with Ki of 0.49 nM in a cell-free assay and also inhibits VEGFR2, Aurora-A, FGFR3, Flt3, Fms (CSF1R), Ret and Yes. It shows ~100-fold selectivity for Chk1 than Chk2. |
|
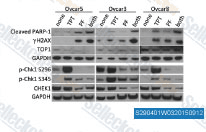
|
| S6385 | Prexasertib (LY2606368) |
Prexasertib (LY2606368, ACR 368) is a selective ATP competitor inhibitor of Chk1 and Chk2 with IC50s of 1 nM and 8 nM in cell-free assays, respectively. Prexasertib also inhibits RSK1 with an IC50 of 9 nM in cell-free assay. |
|
|
| S8632 | BML-277 (Chk2 Inhibitor II) | BML-277 (Chk2 Inhibitor II) is an ATP-competitive inhibitor of Chk2 with IC50 of 15 nM, and it is 1000-fold more selective toward Chk2 serine/threonine kinase than for Chk1 and Cdk1/B kinases. This compound dose dependently protects human CD4(+) and CD8(+) T-cells from apoptosis due to ionizing radiation. |
|
|
| S8253 | CCT245737 (SRA737) | CCT245737 (SRA737) is an orally active CHK1 inhibitor with The IC50 of 1.4 nM. It exhibits >1,000-fold selectivity against CHK2 and CDK1. |
|
|
| S8526 | GDC-0575 | GDC-0575 is a potent and selective CHK1 inhibitor with an IC50 of 1.2 nM. |
|
|
| E1991 | BBI-355 | BBI-355 is an oral, potent, and selective small molecule inhibitor of CHK1 with an IC50 of 0.3 nM. It demonstrates significant anti-tumor activity as a single agent and in combination with targeted therapies in multiple ecDNA+ oncogene amplified tumor models. | ||
| E1653 | CCT241533 hydrochloride | CCT241533 hydrochloride is an ATP competitive, potent, and selective inhibitor of CHK2 with an IC50 of 3 nM and a Ki of 1.16 nM and shows minimal cross-reactivity against a panel of kinases. CCT241533 inhibits CHK2 activity in human tumor cell lines when subjected to DNA damage. | ||
| S7740 | SAR-020106 | SAR-020106 is an ATP-competitive, potent, and selective CHK1 inhibitor with an IC50 of 13.3 nM. | ||
| E5832New | BBI-2779 | BBI-2779 is a potent, selective, and orally active inhibitor of CHK1 with an IC50 of 0.3 nM. It preferentially kills cancer cells containing extrachromosomal DNA (ecDNA) by inducing replication stress and cell death. In gastric cancer models with FGFR2 gene amplification on ecDNA, BBI-2779 suppresses tumor growth and prevents drug resistance. | ||
| E1667 | LY2880070 | LY2880070 is an orally active inhibitor of CHK1. This compound can be used as an anticancer agent and shows promise in preclinical models of pancreatic ductal adenocarcinoma (PDAC) when used in combination with DNA-damaging agents. | ||
| S8148 | PD0166285 | PD0166285 is a potent Wee1 and Chk1 inhibitor with activity at nanomolar concentrations (IC50=24 nM for Wee1 and 72 nM for Myt1). This compound is also a novel G2 checkpoint abrogator. It induces apoptosis. |
|
|
| S9639 | Gartisertib (M4344, VX-803) | VX-803 (M4344, ATR inhibitor 2) is an ATP-competitive, orally active, and selective inhibitor of ataxia telangiectasia and Rad3 related (ATR) kinase with Ki of < 150 pM. VX-803 (M4344) potently inhibits ATR-driven phosphorylated checkpoint kinase-1 (P-Chk1) phosphorylation with IC50 of 8 nM. VX-803 (M4344) exhibits potential antineoplastic activity. |
|
|
| S6740 | DB07268 | DB07268 is a potent and selective JNK1 inhibitor with an IC50 value of 9 nM and exhibits at least 70- to 90-fold greater potency against JNK1 than CHK1, CK2, and PLK. |
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.