-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
research use only
PD168393 EGFR inhibitor
Cat.No.S7039
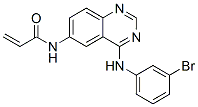
Chemical Structure
Molecular Weight: 369.22
Quality Control
| Related Targets | VEGFR PDGFR FGFR c-Met Src MEK CSF-1R FLT3 HER2 c-Kit |
|---|---|
| Other EGFR Inhibitors | Lazertinib (YH25448) Icotinib Hydrochloride Sunvozertinib AG-490 AG-1478 Canertinib (CI-1033) Rociletinib (CO-1686) WZ4002 Poziotinib (NOV120101, HM781-36B) Genistein |
Solubility
|
In vitro |
DMSO
: 74 mg/mL
(200.42 mM)
Water : Insoluble Ethanol : Insoluble |
Molarity Calculator
|
In vivo |
|||||
In vivo Formulation Calculator (Clear solution)
Step 1: Enter information below (Recommended: An additional animal making an allowance for loss during the experiment)
Step 2: Enter the in vivo formulation (This is only the calculator, not formulation. Please contact us first if there is no in vivo formulation at the solubility Section.)
Calculation results:
Working concentration: mg/ml;
Method for preparing DMSO master liquid: mg drug pre-dissolved in μL DMSO ( Master liquid concentration mg/mL, Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug. )
Method for preparing in vivo formulation: Take μL DMSO master liquid, next addμL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O, mix and clarify.
Method for preparing in vivo formulation: Take μL DMSO master liquid, next add μL Corn oil, mix and clarify.
Note: 1. Please make sure the liquid is clear before adding the next solvent.
2. Be sure to add the solvent(s) in order. You must ensure that the solution obtained, in the previous addition, is a clear solution before proceeding to add the next solvent. Physical methods such
as vortex, ultrasound or hot water bath can be used to aid dissolving.
Chemical Information, Storage & Stability
| Molecular Weight | 369.22 | Formula | C17H13BrN4O |
Storage (From the date of receipt) | |
|---|---|---|---|---|---|
| CAS No. | 194423-15-9 | Download SDF | Storage of Stock Solutions |
|
|
| Synonyms | N/A | Smiles | C=CC(=O)NC1=CC2=C(C=C1)N=CN=C2NC3=CC(=CC=C3)Br | ||
Mechanism of Action
| Features |
Preclinical compound used in the design of CI-1033.
|
|---|---|
| Targets/IC50/Ki |
EGFR
0.70 nM
|
| In vitro |
PD 168393 is docked into the ATP binding pocket of EGFR TK. This compound completely suppresses EGF-dependent receptor autophosphorylation in A431 cells during continuous exposure, with continous suppression even after 8 hr in compound-free medium. It inhibits heregulin-induced tyrosine phosphorylation in MDA-MB-453 cells with IC50 of 5.7 nM. This chemical is inactive against insulin, PDGF and basic FGFR TKs as well as PKC. It inhibits EGF-mediated tyrosine phosphorylation in HS-27 human fibroblasts with IC50 of 1-6 nM but has little effect on FGF- or PDGF-mediated tyrosine phosphorylation. This compound shows rapid and potent inhibition of Her2-induced tyrosine phosphorylation with IC50 of ~100 nM in 3T3-Her2 cells. It also inhibits phosphorylation of PLCγ1/Stat1/Dok1/δ-catenin in 3T3-Her2 cells, except for Fyb.
|
| In vivo |
PD 168393 produces tumor growth inhibition of 115% in A431 human epidermoid carcinoma xenograft in nude mice, with 50% reduced phosphotyrosine content of EGFR. This compound also shows a low plasma concentration.
|
References |
|
Tech Support
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
Signaling Pathway Map
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.