|
How to Cite 1. For In-Text Citation (Materials & Methods): 2. For Key Resources Table: |
||
|
Toll Free: (877) 796-6397 -- USA and Canada only -- |
Fax: +1-832-582-8590 Orders: +1-832-582-8158 |
Tech Support: +1-832-582-8158 Ext:3 Please provide your Order Number in the email. We strive to reply to |
Technical Data
| Formula | C26H31Cl2N7O3 |
||||||
| Molecular Weight | 560.48 | CAS No. | 872511-34-7 | ||||
| Solubility (25°C)* | In vitro | DMSO | 27 mg/mL (48.17 mM) | ||||
| Water | Insoluble | ||||||
| Ethanol | Insoluble | ||||||
| In vivo (Add solvents to the product individually and in order) |
|
||||||
|
* <1 mg/ml means slightly soluble or insoluble. * Please note that Selleck tests the solubility of all compounds in-house, and the actual solubility may differ slightly from published values. This is normal and is due to slight batch-to-batch variations. * Room temperature shipping (Stability testing shows this product can be shipped without any cooling measures.) |
|||||||
Preparing Stock Solutions
Biological Activity
| Description | Infigratinib (BGJ398) is a potent and selective FGFR inhibitor for FGFR1/2/3 with IC50 of 0.9 nM/1.4 nM/1 nM in cell-free assays, >40-fold selective for FGFR versus FGFR4 and VEGFR2, and little activity to Abl, Fyn, Kit, Lck, Lyn and Yes. Phase 2. | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Targets |
|
||||||||||
| In vitro | Infigratinib (BGJ398) also prevents VEGFR2 with low potency, with an IC50 of 0.18 μM for inhibiting this target. It suppresses other kinases including ABL, FYN, KIT, LCK, LYN and YES with IC50 values of 2.3 μM, 1.9 μM, 0.75 μM, 2.5 μM, 0.3 μM and 1.1 μM, respectively. At the cellular level, this compound inhibits the proliferation of the FGFR1-, FGFR2-Q, and FGFR3-dependent BaF3 cells with IC50 of 2.9 μM, 2.0 μM and 2 μM, respectively. It interferes with autophosphorylation on specific tyrosine residues including FGFR-WT, FGFR2-WT, FGFR3-K650E, FGFR3-S249C and FGFR4-WT with IC50 of 4.6 nM, 4.9 nM, 5 nM, 5 nM and 168 nM, respectively. Additionally, it suppresses proliferation of the cancer cells with wild-type (WT) FGFR3 overexpression such as RT112, RT4, SW780 and JMSU1 with IC50 of 5 nM, 30 nM, 32 nM and 15 nM, respectively. | ||||||||||
| In vivo | In this orthotopic xenograft bladder cancer model, BGJ398 induces tumor growth inhibition and stasis after oral administration for 12 consecutive days at the doses of 10 and 30 mg/kg, respectively. Interestingly, the animals that received this compound exhibit either no body weight loss (10 mg/kg) or 10% body weight gain (30 mg/kg), a further indication of efficacy. RT112 tumor-bearing and female Rowett rats receive a single oral administration of the monophosphate salt of Infigratinib (BGJ398) at the doses of 4.25 and 8.51 mg/kg. It significantly decreases the levels of pFRS2 and pMAPK in a dose-dependent manner. Additionally, it inhibits significantly bFGF-stimulated angiogenesis in a dose-dependent manner. However, it does not impair VEGF-induced blood vessel formation. |
Protocol (from reference)
| Kinase Assay: [1] |
|
|---|---|
| Cell Assay:[1] |
|
| Animal Study:[1] |
|
References
|
Customer Product Validation

-
Data from [ Hepatology , 2014 , 59(4), 1427-34 ]
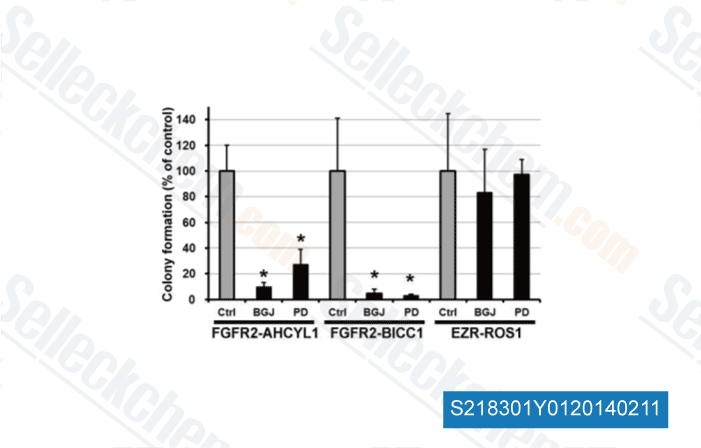
-
Data from [ Hepatology , 2014 , 59(4), 1427-34 ]

-
Data from [ J Cell Physiol , 2014 , 229(11), 1647-59 ]

-
, , Oncogene, 2017, 36(27):3831-3841
Selleck's BGJ398 (Infigratinib) Has Been Cited by 257 Publications
| High-Purity Functional Corneal Endothelial Cells From Human Induced Pluripotent Stem Cells via a Novel Wash-Out Method [ MedComm (2020), 2026, 7(4):e70650] | PubMed: 41930340 |
| 3D-Printed Scaffolds Promote Enhanced Spinal Organoid Formation for Use in Spinal Cord Injury [ Adv Healthc Mater, 2025, e04817.] | PubMed: 40702833 |
| Selective degradation of FGFR1/2 overcomes antiestrogen resistance in ER+ breast cancer with FGFR1/2 alterations [ Cancer Lett, 2025, 619:217668] | PubMed: 40127812 |
| Abrogation of FGFR signaling blocks β-catenin-induced adrenocortical hyperplasia and aldosterone production [ JCI Insight, 2025, 10(21)e184863] | PubMed: 40956622 |
| Fibroblast growth factor receptor signaling modulates cholesterol storage in a SOAT1-dependent manner to promote mammary tumor cell invasion [ Breast Cancer Res, 2025, 27(1):132] | PubMed: 40665359 |
| Exploiting collateral sensitivity in the evolution of resistance to tyrosine kinase inhibitors in soft tissue sarcomas [ Commun Biol, 2025, 8(1):1185] | PubMed: 40781553 |
| Sonic hedgehog and fibroblast growth factor 8 regulate the evolution of amniote facial proportions [ Commun Biol, 2025, 8(1):84] | PubMed: 39827295 |
| Aspirin-triggered DHA metabolites inhibit angiogenesis [ Front Pharmacol, 2025, 16:1524980] | PubMed: 40070577 |
| Generation of an induced pluripotent stem cell line (NCHi023-A) from a 41-year-old male with nemaline myopathy carrying autosomal dominant ACTA1 c.809-10C>A mutation [ Stem Cell Res, 2025, 85:103701] | PubMed: 40147060 |
| Characterization of an induced pluripotent stem cell line NCHi025-A from a 1-year-old female with pulmonary stenosis harboring a heterozygous HAND2 variant [ Stem Cell Res, 2025, 86:103733] | PubMed: 40378586 |
RETURN POLICY
Selleck Chemical’s Unconditional Return Policy ensures a smooth online shopping experience for our customers. If you are in any way unsatisfied with your purchase, you may return any item(s) within 7 days of receiving it. In the event of product quality issues, either protocol related or product related problems, you may return any item(s) within 365 days from the original purchase date. Please follow the instructions below when returning products.
SHIPPING AND STORAGE
Selleck products are transported at room temperature. If you receive the product at room temperature, please rest assured, the Selleck Quality Inspection Department has conducted experiments to verify that the normal temperature placement of one month will not affect the biological activity of powder products. After collecting, please store the product according to the requirements described in the datasheet. Most Selleck products are stable under the recommended conditions.
NOT FOR HUMAN, VETERINARY DIAGNOSTIC OR THERAPEUTIC USE.