-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
research use only
Phospho-Histone H2A (Ser129) Antibody [A4N9]
Cat.No.: F3871
Application:
Reactivity:
-
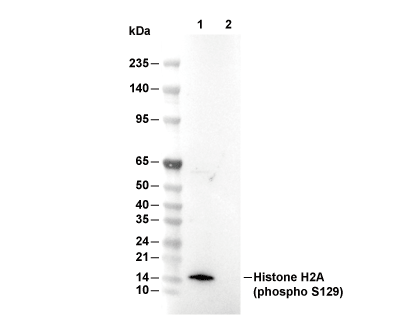 Lane 1: Saccharomyces cerevisiae (0.2% Methyl methanesulfonate, 1 h), Lane 2: Saccharomyces cerevisiae
Lane 1: Saccharomyces cerevisiae (0.2% Methyl methanesulfonate, 1 h), Lane 2: Saccharomyces cerevisiae
Experiment Essentials
WB
Recommended SDS-PAGE separating gel concentration: 20%.
Recommended wet transfer conditions: 200 mA, 60 min,Recommended to use 0.22 μm PVDF membrane.
Exposure time of at least 150s is recommended.
Recommended SDS-PAGE separating gel concentration: 20%.
Recommended wet transfer conditions: 200 mA, 60 min,Recommended to use 0.22 μm PVDF membrane.
Exposure time of at least 150s is recommended.
Usage Information
| Dilution |
|---|
|
| Application |
|---|
| WB, ChIP, ELISA |
| Reactivity |
|---|
| Human, Saccharomyces cerevisiae |
| Source |
|---|
| Rabbit Monoclonal Antibody |
| Storage Buffer |
|---|
| PBS, pH 7.2+50% Glycerol+0.05% BSA+0.01% NaN3 |
| Storage (from the date of receipt) |
|---|
| -20°C (avoid freeze-thaw cycles), 2 years |
| Predicted MW Observed MW |
|---|
| 14 kDa 14 kDa |
| *Why do the predicted and actual molecular weights differ? The following reasons may explain differences between the predicted and actual protein molecular weight. Post-translational modifications(e.g., phosphorylation, glycosylation); Splice variants and isoforms; Relative charge; Multimerization. |
| Positive Control | Saccharomyces cerevisiae cells; Saccharomyces cerevisiae (Methyl methanesulfonate, 0.2%, 1 h) |
|---|---|
| Negative Control |
Experimental Methods
| WB |
|---|
Experimental Protocol:
Sample preparation
1. Tissue: Lyse the tissue sample by adding an appropriate volume of ice-cold RIPA/NP-40 Lysis Buffer (containing Protease Inhibitor Cocktail, Phosphatase Inhibitor Cocktail),and homogenize the tissue at a low temperature. 2. Adherent cell: Aspirate the culture medium and wash the cells with ice-cold PBS twice. Lyse the cells by adding an appropriate volume of RIPA/NP-40 Lysis Buffer (containing Protease Inhibitor Cocktail, Phosphatase Inhibitor Cocktail) and put the sample on ice for 5 min. 3. Suspension cell: Transfer the culture medium to a pre-cooled centrifuge tube. Centrifuge and aspirate the supernatant. Wash the cells with ice-cold PBS twice. Lyse the cells by adding an appropriate volume of RIPA/NP-40 Lysis Buffer (containing Protease Inhibitor Cocktail, Phosphatase Inhibitor Cocktail) and put the sample on ice for 5 min. 4. Place the lysate into a pre-cooled microcentrifuge tube. Centrifuge at 4°C for 15 min. Collect the supernatant;
5. Remove a small volume of lysate to determine the protein concentration;
6. Combine the lysate with protein loading buffer. Boil 20 µL sample under 95-100°C for 5 min. Centrifuge for 5 min after cool down on ice.
Electrophoretic separation
1. According to the concentration of extracted protein, load appropriate amount of protein sample and marker onto SDS-PAGE gels for electrophoresis. Recommended separating gel (lower gel) concentration: 20%. Reference Table for Selecting SDS-PAGE Separation Gel Concentrations 2. Power up 80V for 30 minutes. Then the power supply is adjusted (110 V~150 V), the Marker is observed, and the electrophoresis can be stopped when the indicator band of the predyed protein Marker where the protein is located is properly separated. (Note that the current should not be too large when electrophoresis, too large current (more than 150 mA) will cause the temperature to rise, affecting the result of running glue. If high currents cannot be avoided, an ice bath can be used to cool the bath.)
Transfer membrane
1. Take out the converter, soak the clip and consumables in the pre-cooled converter;
2. Activate PVDF membrane with methanol for 1 min and rinse with transfer buffer;
3. Install it in the order of "black edge of clip - sponge - filter paper - filter paper - glue -PVDF membrane - filter paper - filter paper - sponge - white edge of clip"; 4. The protein was electrotransferred to PVDF membrane. ( 0.22 µm PVDF membrane is recommended )) Reference Table for Selecting PVDF Membrane Pore Size Specifications Recommended conditions for wet transfer: 200 mA, 60 min. ( Note that the transfer conditions can be adjusted according to the protein size. For high-molecular-weight proteins, a higher current and longer transfer time are recommended. However, ensure that the transfer tank remains at a low temperature to prevent gel melting.)
Block
1. After electrotransfer, wash the film with TBST at room temperature for 5 minutes;
2. Incubate the film in the blocking solution ( recommending 5% BSA solution)
for 1 hour at room temperature;
3. Wash the film with TBST for 3 times, 5 minutes each time.
Antibody incubation
1. Use 5% skim milk powder to prepare the primary antibody working liquid (recommended dilution ratio for primary antibody 1:5000), gently shake and incubate with the film at 4°C overnight; 2. Wash the film with TBST 3 times, 5 minutes each time;
3. Add the secondary antibody to the blocking solution and incubate with the film gently at room temperature for 1 hour;
4. After incubation, wash the film with TBST 3 times for 5 minutes each time.
Antibody staining
1. Add the prepared ECL luminescent substrate (or select other color developing substrate according to the second antibody) and mix evenly;
2. Incubate with the film for 1 minute, remove excess substrate (keep the film moist), wrap with plastic film, and expose in the imaging system. (Exposure time of at least 150s is recommended) |
Biological Description
| Specificity |
|---|
| Phospho-Histone H2A (Ser129) Antibody [A4N9] detects endogenous levels of total Histone H2A protein only when it is phosphorylated at Ser129. |
| Subcellular Location |
|---|
| Chromosome, Nucleosome core, Nucleus |
| Uniprot ID |
|---|
| P04912 |
| Clone |
|---|
| A4N9 |
| Synonym(s) |
|---|
| H2A2; YBL003C; YBL0103; HTA2; Histone H2A.2 |
| Background |
|---|
| Phospho Histone H2A at Ser129 (H2A S129ph) is a conserved, DNA damage–induced histone mark that plays a central role in the chromatin based signaling cascade coordinating double strand break (DSB) repair and genome integrity, particularly in yeast and mammalian systems where it functions as part of the broader H2A.X family of variant histones. The modification occurs at a C terminal SQ motif that is phosphorylated by phosphoinositide 3 kinase related kinases such as ATM and ATR in response to DNA breaks, generating a localized phospho epitope that spreads over kilobase scale chromatin regions flanking the lesion and thereby serving as a nucleation platform for downstream repair factors. H2A S129ph directly recruits chromatin modifying complexes such as the NuA4 histone acetyltransferase and Ino80/Swr1 type ATP dependent remodeling complexes, which are anchored through specific subunits that recognize the phosphorylated tail, thus promoting acetylation and restructuring of nearby nucleosomes to enhance accessibility of damaged DNA to repair enzymes. This phospho H2A–dependent recruitment supports efficient homologous recombination and non homologous end joining by facilitating the assembly and retention of DSB sensing and processing factors at the break site, while also interfacing with cell cycle checkpoints to delay progression until repair is complete. H2A S129ph is widely used as a sensitive, high resolution readout for DSB generation and repair efficiency, and dysregulation of its phosphorylation or recognition machinery contributes to defective repair responses, increased genomic instability, and hypersensitivity to genotoxic agents, so that H2A S129ph dependent signaling is ultimately dysregulated in DNA repair deficient and cancer prone backgrounds. |
| References |
|---|
|
Tech Support
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.