-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
research use only
IL-1β Antibody [N17M3]
Cat.No.: F4785
Application:
Reactivity:
-
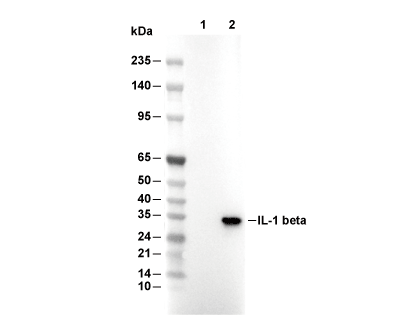 Lane 1: THP-1, Lane 2: THP-1 (LPS, 100ng/ml, 3 h)
Lane 1: THP-1, Lane 2: THP-1 (LPS, 100ng/ml, 3 h)
Usage Information
| Dilution |
|---|
|
| Application |
|---|
| WB, IP, IHC, IF, FCM |
| Reactivity |
|---|
| Mouse, Rat, Human |
| Source |
|---|
| Rabbit Monoclonal Antibody |
| Storage Buffer |
|---|
| PBS, pH 7.2+50% Glycerol+0.05% BSA+0.01% NaN3 |
| Storage (from the date of receipt) |
|---|
| -20°C (avoid freeze-thaw cycles), 2 years |
| Predicted MW Observed MW |
|---|
| 31 kDa 28 kDa,31 kDa |
| *Why do the predicted and actual molecular weights differ? The following reasons may explain differences between the predicted and actual protein molecular weight. Post-translational modifications(e.g., phosphorylation, glycosylation); Splice variants and isoforms; Relative charge; Multimerization. |
Biological Description
| Specificity |
|---|
| IL-1β Antibody [N17M3] detects endogenous levels of total IL-1β protein. |
| Clone |
|---|
| N17M3 |
| Synonym(s) |
|---|
| IL1F2, IL1B, Interleukin-1 beta, IL-1 beta, Catabolin |
| Background |
|---|
| Interleukin-1 beta (IL-1β) belongs to the interleukin-1 cytokine family comprising eleven members and functions as one of the most potent proinflammatory cytokines orchestrating innate immune responses and resistance to pathogens while also exacerbating damage during chronic disease and acute tissue injury. IL-1β synthesis occurs initially as an inactive precursor termed pro-IL-1β in response to pathogen-associated molecular patterns (PAMPs) recognized by pattern recognition receptors including Toll-like receptors, which induce transcriptional upregulation through NF-κB activation in cells predominantly of the innate immune system such as monocytes, macrophages, and dendritic cells, although neutrophils, epithelial cells, and fibroblasts also produce IL-1β under specific stimuli. The protein lacks a signal peptide for conventional ER-Golgi secretion and instead undergoes proteolytic processing by caspase-1, which cleaves pro-IL-1β into the biologically active mature cytokine following assembly of multi-protein inflammasome complexes including NLRP3, NLRC4, NLRP1, and AIM2 that respond to secondary PAMPs or danger-associated molecular patterns (DAMPs) such as extracellular ATP, nigericin, monosodium urate crystals, silica, cholesterol crystals, beta-amyloid aggregates, and bacterial infection. Mature IL-1β binds the interleukin-1 receptor type I (IL-1RI) on target cells, recruiting the accessory protein IL-1RAcP to form a signaling-competent receptor complex that triggers MyD88-dependent assembly of intracellular signaling platforms involving IRAK family kinases and TRAF6, culminating in activation of NF-κB, JNK, and p38 mitogen-activated protein kinase pathways through combinatorial phosphorylation and ubiquitination events. These signaling cascades cooperatively induce transcription of canonical IL-1 target genes including IL-6, IL-8, monocyte chemoattractant protein-1 (MCP-1), cyclooxygenase-2 (COX-2), IκBα, IL-1α, IL-1β itself, and MAP kinase phosphatase-1 (MKP-1) through both transcriptional activation and posttranscriptional mechanisms, rapidly amplifying inflammatory responses. IL-1β secretion proceeds through non-conventional pathways independent of the ER-Golgi route, with multiple distinct mechanisms operating on a continuum depending on stimulus strength and extracellular IL-1β requirement—these include rescue and redirection from autophagosomes targeted for lysosomal degradation that can be diverted to lysosome exocytosis, protected release within shed microvesicles or exosomes that safeguard IL-1β for delivery to distant sites given its short plasma half-life, and terminal release during pyroptosis wherein caspase-1-dependent pore formation in the plasma membrane enables IL-1β passage followed by osmotic lysis. IL-1β executes pleiotropic biological functions including acting as an endogenous pyrogen inducing fever through hypothalamic signaling, stimulating prostaglandin synthesis, promoting neutrophil influx and activation, triggering T-cell activation with specific enhancement of Th17 differentiation, synergizing with IL-12 to induce interferon-gamma synthesis from Th1 cells, activating B-cells and antibody production, stimulating fibroblast proliferation and collagen synthesis, and inducing vascular endothelial growth factor (VEGF) production synergistically with TNF and IL-6 to promote angiogenesis. The cytokine participates in transduction of inflammation downstream of pyroptosis wherein mature IL-1β specifically passes through gasdermin-D (GSDMD) pores formed during this lytic cell death pathway, enabling extracellular release to amplify inflammatory signaling. IL-1β activity becomes restricted at three regulatory levels—synthesis and release controlled by inflammasome activation thresholds, membrane receptor availability modulated by the endogenous IL-1 receptor antagonist (IL-1RA) that competitively blocks IL-1RI without triggering signaling, and intracellular signal transduction subject to negative feedback through MKP-1-mediated dephosphorylation of MAPKs and IκBα-mediated sequestration of NF-κB. Dysregulated IL-1β production underlies numerous autoinflammatory diseases, with therapeutic blockade using IL-1RA or neutralizing antibodies demonstrating efficacy in conditions including gout triggered by urate crystal-induced NLRP3 activation, atherosclerosis exacerbated by cholesterol crystal inflammasome activation, type 2 diabetes associated with metabolic inflammation, rheumatoid arthritis, and cryopyrin-associated periodic syndromes caused by gain-of-function NLRP3 mutations. |
| References |
|---|
|
Tech Support
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.