-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
research use only
IGFBP3 Antibody [G15L3]
Cat.No.: F9208
Application:
Reactivity:
-
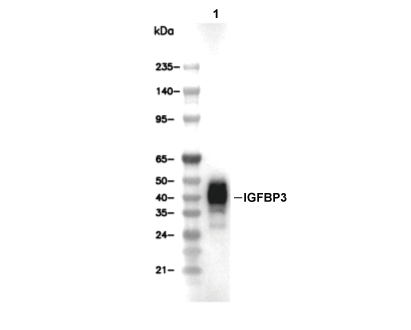 Lane 1: AsPC-1
Lane 1: AsPC-1
Usage Information
| Dilution |
|---|
|
| Application |
|---|
| WB, IP, IHC, IF |
| Reactivity |
|---|
| Human |
| Source |
|---|
| Rabbit Monoclonal Antibody |
| Storage Buffer |
|---|
| PBS, pH 7.2+50% Glycerol+0.05% BSA+0.01% NaN3 |
| Storage (from the date of receipt) |
|---|
| -20°C (avoid freeze-thaw cycles), 2 years |
| Predicted MW |
|---|
| 19 kDa |
Biological Description
| Specificity |
|---|
| IGFBP3 Antibody [G15L3] detects endogenous levels of total IGFBP3 protein. |
| Clone |
|---|
| G15L3 |
| Synonym(s) |
|---|
| acid stable subunit of the 140 K IGF complex; binding protein 29; binding protein 53; BP-53; IBP-3; IBP3; IGF-binding protein 3; IGFBP-3; IGFBP3; insulin like growth factor binding protein 3 |
| Background |
|---|
| Insulin-like growth factor binding protein-3 (IGFBP3) represents the most abundant member of the six-member IGFBP family in human circulation, functioning primarily as the major carrier protein for insulin-like growth factors IGF-I and IGF-II within ternary complexes containing the acid-labile subunit, where it prolongs IGF half-life from minutes to over twelve hours while regulating IGF bioavailability through high-affinity sequestration. IGFBP3 comprises three structural domains—a highly conserved cysteine-rich N-terminal domain containing twelve cysteine residues forming six intradomain disulfide bridges, a non-conserved central linker domain, and a conserved cysteine-rich C-terminal domain with six cysteines forming three disulfide bridges—with both terminal domains contributing cooperatively to IGF binding through distinct contact residues that together generate nanomolar binding affinity. The protein undergoes post-translational modifications, including N-glycosylation at three asparagine residues (Asn89, Asn109, Asn172) contributing variable carbohydrate content, producing characteristic doublets detectable by Western blot, phosphorylation at multiple serine residues by casein kinase 2 and DNA-dependent protein kinase, altering glycosaminoglycan binding and IGF affinity, and proteolytic cleavage by matrix metalloproteinases and pregnancy-associated plasma protein-A2 reducing IGF binding affinity to release bioactive IGF for receptor activation. IGFBP3 regulates IGF-dependent cellular processes through multiple mechanisms—canonical inhibition of IGF1R signaling by sequestering IGF-I and IGF-II preventing receptor access, potentiation of IGF1R activation through interaction with cell-surface glycosaminoglycans and extracellular matrix proteins concentrating IGFs near receptors, and suppression of IGF1R signaling by activating protein phosphatase 2A following binding to transforming growth factor-β receptor type V (TGFβRV/LRP1), leading to dephosphorylation of Akt and Ras/MAPK pathway components. The protein executes extensive IGF-independent functions initiated at the cell surface through interactions with transmembrane protein 219 (TMEM219), TGFβRI, TGFβRII, and TGFβRV receptors—TMEM219 binding triggers caspase-8-dependent apoptosis and forms calcium/calmodulin-dependent complexes inducing autophagy, while TGFβRV ligation activates protein phosphatase 2A, downregulating Akt survival pathways. IGFBP3 possesses cell-penetrating peptide properties mediated by overlapping glycosaminoglycan-binding, nuclear localization signal, and metal-binding domains within the C-terminal region, enabling cellular uptake through clathrin-mediated endocytosis via transferrin receptor interaction, caveolae-mediated endocytosis involving caveolin-1, and fluid-phase pinocytosis, with heparan sulfate proteoglycans facilitating membrane association and internalization. Following internalization, IGFBP3 translocates to the nucleus through direct importin-β binding independent of importin-α adaptor protein in an ATP- and GTP-dependent manner, where the bipartite nuclear localization signal within the C-terminal domain directs nuclear accumulation regulated by polyubiquitination and proteasomal degradation. Nuclear IGFBP3 functions as a transcriptional regulator through interactions with class II nuclear hormone receptors, including retinoid X receptor-α and vitamin D receptor, modulating their transcriptional activity, binds to DNA-dependent protein kinase catalytic subunit, participating in non-homologous end-joining double-strand DNA repair pathways, and interacts with numerous transcription factors influencing gene expression programs controlling apoptosis, cell cycle progression, and metabolic homeostasis. IGFBP3 regulates autophagy through multiple mechanisms—hypoglycosylated IGFBP3 under glucose and oxygen deprivation binds glucose-regulated protein 78 (GRP78) inducing autophagic survival responses in nutrient-stressed breast cancer cells, TMEM219-calmodulin-calcium/calmodulin-dependent kinase II complex formation triggers autophagy in kidney epithelial cells, interleukin-13-induced autophagy in bronchial epithelial cells depends on IGFBP3, while in corneal epithelium IGFBP3 suppresses mitophagy maintaining mitochondrial function through regulation of BNIP3L/NIX mitophagy receptor expression with nuclear accumulation correlating with altered mitochondrial respiration and cristae morphology. The protein modulates sphingolipid metabolism by upregulating sphingosine kinase 1 activity, generating pro-survival sphingosine 1-phosphate while decreasing pro-apoptotic ceramide levels, potentiating ligand-dependent activation of both IGF1R and EGFR through sphingosine 1-phosphate receptor signaling with EGFR transactivating IGF1R independent of IGF binding. IGFBP3 exhibits context-dependent pro-apoptotic or anti-apoptotic functions—the protein induces apoptosis in prostate cancer cells through caspase-dependent mechanisms operating independently of IGF binding, secretion, or nuclear concentration, activates STAT-1 and inhibits Akt/IGF-1R survival pathways in breast cancer cells, promoting apoptosis, yet paradoxically high nuclear IGFBP3 in triple-negative breast cancer xenografts correlates negatively with cleaved caspase-3 and predicts shorter survival with tumor growth driven by IGFBP3-stimulated sphingosine kinase 1 and EGFR activation. IGFBP3 expression becomes regulated at transcriptional and post-translational levels with disruption implicated in multiple malignancies—reduced circulating IGFBP3 combined with elevated IGF-I correlates with increased breast, colon, lung, and prostate cancer risk, p53 tumor suppressor directly induces IGFBP3 transcription positioning it as a p53-regulated growth inhibitor, IGFBP3 levels decline in malignant cells relative to normal tissues, and elevated nuclear IGFBP3 in prostate cancer significantly predicts tumor recurrence while serving as both prognostic marker and potential therapeutic target. |
| References |
|---|
|
Tech Support
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.