-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
research use only
Gα(q) Antibody [C11L2]
Cat.No.: F7457
Application:
Reactivity:
-
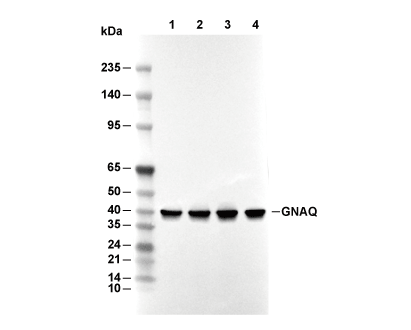 Lane 1: 293T, Lane 2: HeLa, Lane 3: C6, Lane 4: RAW264.7
Lane 1: 293T, Lane 2: HeLa, Lane 3: C6, Lane 4: RAW264.7
Usage Information
| Dilution |
|---|
|
| Application |
|---|
| WB, IP, IHC |
| Reactivity |
|---|
| Mouse, Rat, Human |
| Source |
|---|
| Rabbit Monoclonal Antibody |
| Storage Buffer |
|---|
| PBS, pH 7.2+50% Glycerol+0.05% BSA+0.01% NaN3 |
| Storage (from the date of receipt) |
|---|
| -20°C (avoid freeze-thaw cycles), 2 years |
| Predicted MW Observed MW |
|---|
| 42 kDa 42 kDa |
| *Why do the predicted and actual molecular weights differ? The following reasons may explain differences between the predicted and actual protein molecular weight. Post-translational modifications(e.g., phosphorylation, glycosylation); Splice variants and isoforms; Relative charge; Multimerization. |
Biological Description
| Specificity |
|---|
| Gα(q) Antibody [C11L2] detects endogenous levels of total Gα(q) protein. |
| Clone |
|---|
| C11L2 |
| Synonym(s) |
|---|
| GAQ, GNAQ, Guanine nucleotide-binding protein G(q) subunit alpha, Guanine nucleotide-binding protein alpha-q |
| Background |
|---|
| G protein subunit alpha q (GNAQ) is a ubiquitously expressed heterotrimeric G protein alpha subunit of the Gq/11 family that couples activated G protein–coupled receptors to phospholipase C‑β–dependent phosphoinositide signaling and calcium mobilization, with prominent roles in vascular, hematopoietic, and neural tissues. The protein contains a canonical GTP‑binding domain with switch regions that undergo conformational changes upon GDP–GTP exchange, an N‑terminal segment that contributes to membrane association and interaction with Gβγ, and C‑terminal residues that recognize activated GPCRs and determine receptor coupling specificity. Activation begins when a ligand‑bound seven‑transmembrane receptor promotes GDP release from GNAQ and binding of GTP, which dissociates Gαq from Gβγ and positions the active GTP‑bound alpha subunit to engage downstream effectors, primarily phospholipase C‑β isoforms. Direct activation of PLC‑β by Gαq drives hydrolysis of phosphatidylinositol 4,5‑bisphosphate into inositol 1,4,5‑trisphosphate and diacylglycerol, triggering release of calcium from intracellular stores and activation of protein kinase C, and these second messengers converge on multiple pathways that regulate gene expression, cytoskeletal dynamics, proliferation, secretion, and contractility. Intrinsic GTPase activity of GNAQ, supported by regulatory proteins, converts GTP to GDP to terminate signaling and allows reassociation with Gβγ to reform the inactive heterotrimer, creating a tightly controlled cycle that links receptor occupancy to the duration and amplitude of phosphoinositide and calcium signaling. In vascular development and function, GNAQ participates in GPCR pathways that shape endothelial and smooth‑muscle behavior and contributes to regulation of vessel patterning, tone, and remodeling, while in the immune system it regulates B cell selection and survival, prevents B cell–dependent autoimmunity, and influences chemotaxis of myeloid cells through coupling to chemokine and lipid receptors. Roles in platelet activation arise from Gαq‑mediated coupling of thromboxane and other platelet GPCRs to PLC‑β activation and calcium‑dependent granule release and integrin activation, which makes GNAQ central to hemostatic and thrombotic responses. Pathogenic variants that impair GTP hydrolysis generate a constitutively active Gαq that remains locked in the GTP‑bound state, causing persistent downstream signaling with sustained PLC‑β activation, chronic calcium elevation, and PKC–MAPK pathway engagement. Somatic mutations affecting conserved residues such as Q209 and R183 occur frequently in uveal melanoma, blue nevi, and related melanocytic lesions, where constitutive GNAQ activation serves as an early oncogenic driver that promotes MAPK and YAP‑dependent transcriptional programs linked to proliferation and survival. Mosaic activating mutations in GNAQ underlie Sturge–Weber syndrome, in which chronic Gαq signaling disrupts normal regulation of blood vessel development and leads to abnormal capillary malformations, leptomeningeal vascular anomalies, and associated neurologic manifestations. |
| References |
|---|
|
Tech Support
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.