-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
research use only
CaMKIIδ Antibody (Rabbit mAb) [E8P2]
Cat.No.: F2409
Application:
Reactivity:
-
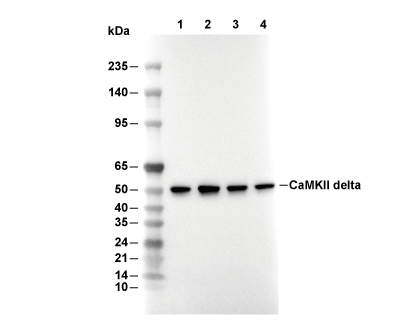 Lane 1: Hela, Lane 2: SW480, Lane 3: A431, Lane 4: Mouse heart
Lane 1: Hela, Lane 2: SW480, Lane 3: A431, Lane 4: Mouse heart
Usage Information
| Dilution |
|---|
|
| Application |
|---|
| WB, IHC |
| Reactivity |
|---|
| Mouse, Rat, Human |
| Source |
|---|
| Rabbit Monoclonal Antibody |
| Storage Buffer |
|---|
| PBS, pH 7.2+50% Glycerol+0.05% BSA+0.01% NaN3 |
| Storage (from the date of receipt) |
|---|
| -20°C (avoid freeze-thaw cycles), 2 years |
| Predicted MW Observed MW |
|---|
| 56 kDa 56 kDa,36 kDa |
| *Why do the predicted and actual molecular weights differ? The following reasons may explain differences between the predicted and actual protein molecular weight. Post-translational modifications(e.g., phosphorylation, glycosylation); Splice variants and isoforms; Relative charge; Multimerization. |
| Positive Control | Human thyroid carcinoma tissue; Human cardiac muscle tissue; Mouse heart tissue; Human skeletal muscle tissue; Rat spleen tissue; Rat heart tissue; Wild-type HAP1 cells; HeLa cells; A431 cells; SW480 cells; SiHa cells; HEK-293T cells |
|---|---|
| Negative Control |
Experimental Methods
| WB |
|---|
Experimental Protocol:
Sample preparation
1. Tissue: Lyse the tissue sample by adding an appropriate volume of ice-cold RIPA/NP-40 Lysis Buffer (containing Protease Inhibitor Cocktail),and homogenize the tissue at a low temperature or lyse it by sonication on ice, then incubate on ice for 30 minutes. 2. Adherent cell: Aspirate the culture medium and wash the cells with ice-cold PBS twice. Lyse the cells by adding an appropriate volume of RIPA/NP-40 Lysis Buffer (containing Protease Inhibitor Cocktail) , sonicate to lyse the cells, and incubate on ice for 30 minutes. 3. Suspension cell: Transfer the culture medium to a pre-cooled centrifuge tube. Centrifuge and aspirate the supernatant. Wash the cells with ice-cold PBS twice. Lyse the cells by adding an appropriate volume of RIPA/NP-40 Lysis Buffer (containing Protease Inhibitor Cocktail) , sonicate to lyse the cells, and incubate on ice for 30 minutes. 4. Place the lysate into a pre-cooled microcentrifuge tube. Centrifuge at 4°C for 15 min. Collect the supernatant;
5. Remove a small volume of lysate to determine the protein concentration;
6. Combine the lysate with protein loading buffer. Boil 20 µL sample under 95-100°C for 5 min. Centrifuge for 5 min after cool down on ice.
Electrophoretic separation
1. According to the concentration of extracted protein, load appropriate amount of protein sample and marker onto SDS-PAGE gels for electrophoresis. Recommended separating gel (lower gel) concentration: 10%. Reference Table for Selecting SDS-PAGE Separation Gel Concentrations 2. Power up 80V for 30 minutes. Then the power supply is adjusted (110 V~150 V), the Marker is observed, and the electrophoresis can be stopped when the indicator band of the predyed protein Marker where the protein is located is properly separated. (Note that the current should not be too large when electrophoresis, too large current (more than 150 mA) will cause the temperature to rise, affecting the result of running glue. If high currents cannot be avoided, an ice bath can be used to cool the bath.)
Transfer membrane
1. Take out the converter, soak the clip and consumables in the pre-cooled converter;
2. Activate PVDF membrane with methanol for 1 min and rinse with transfer buffer;
3. Install it in the order of "black edge of clip - sponge - filter paper - filter paper - glue -PVDF membrane - filter paper - filter paper - sponge - white edge of clip"; 4. The protein was electrotransferred to PVDF membrane. ( 0.45 µm PVDF membrane is recommended ) Reference Table for Selecting PVDF Membrane Pore Size Specifications Recommended conditions for wet transfer: 200 mA, 120 min. ( Note that the transfer conditions can be adjusted according to the protein size. For high-molecular-weight proteins, a higher current and longer transfer time are recommended. However, ensure that the transfer tank remains at a low temperature to prevent gel melting.)
Block
1. After electrotransfer, wash the film with TBST at room temperature for 5 minutes;
2. Incubate the film in the blocking solution for 1 hour at room temperature;
3. Wash the film with TBST for 3 times, 5 minutes each time.
Antibody incubation
1. Use 5% skim milk powder to prepare the primary antibody working liquid (recommended dilution ratio for primary antibody 1:1000), gently shake and incubate with the film at 4°C overnight; 2. Wash the film with TBST 3 times, 5 minutes each time;
3. Add the secondary antibody to the blocking solution and incubate with the film gently at room temperature for 1 hour;
4. After incubation, wash the film with TBST 3 times for 5 minutes each time.
Antibody staining
1. Add the prepared ECL luminescent substrate (or select other color developing substrate according to the second antibody) and mix evenly;
2. Incubate with the film for 1 minute, remove excess substrate (keep the film moist), wrap with plastic film, and expose in the imaging system. |
| IHC |
|---|
Experimental Protocol:
Deparaffinization/Rehydration
1. Deparaffinize/hydrate sections:
2. Incubate sections in three washes of xylene for 5 min each.
3. Incubate sections in two washes of 100% ethanol for 10 min each.
4. Incubate sections in two washes of 95% ethanol for 10 min each.
5. Wash sections two times in dH2O for 5 min each.
6.Antigen retrieval: For Citrate: Heat slides in a microwave submersed in 1X citrate unmasking solution until boiling is initiated; continue with 10 min at a sub-boiling temperature (95°-98°C). Cool slides on bench top for 30 min.
Staining
1. Wash sections in dH2O three times for 5 min each.
2. Incubate sections in 3% hydrogen peroxide for 10 min.
3. Wash sections in dH2O two times for 5 min each.
4. Wash sections in wash buffer for 5 min.
5. Block each section with 100–400 µl of blocking solution for 1 hr at room temperature.
6. Remove blocking solution and add 100–400 µl primary antibody diluent in to each section. Incubate overnight at 4°C.
7. Remove antibody solution and wash sections with wash buffer three times for 5 min each.
8. Cover section with 1–3 drops HRPas needed. Incubate in a humidified chamber for 30 min at room temperature.
9. Wash sections three times with wash buffer for 5 min each.
10. Add DAB Chromogen Concentrate to DAB Diluent and mix well before use.
11. Apply 100–400 µl DAB to each section and monitor closely. 1–10 min generally provides an acceptable staining intensity.
12. Immerse slides in dH2O.
13. If desired, counterstain sections with hematoxylin.
14. Wash sections in dH2O two times for 5 min each.
15. Dehydrate sections: Incubate sections in 95% ethanol two times for 10 sec each; Repeat in 100% ethanol, incubating sections two times for 10 sec each; Repeat in xylene, incubating sections two times for 10 sec each.
16. Mount sections with coverslips and mounting medium.
|
Biological Description
| Specificity |
|---|
| CaMKIIδ Antibody (Rabbit mAb) [E8P2] detects endogenous levels of total CaMKIIδ protein. |
| Subcellular Location |
|---|
| Cell membrane, Membrane, Sarcoplasmic reticulum |
| Uniprot ID |
|---|
| Q13557 |
| Clone |
|---|
| E8P2 |
| Synonym(s) |
|---|
| CAMKD, CAMK2D, Calcium/calmodulin-dependent protein kinase type II subunit delta, CaM kinase II subunit delta, CaMK-II subunit delta |
| Background |
|---|
| CaMKIIδ is the predominant Ca²⁺/calmodulin-dependent protein kinase II isoform in heart that assembles into multimeric holoenzymes and functions as a central integrator of Ca²⁺, β‑adrenergic, and redox signals, translating repetitive Ca²⁺ transients into sustained phosphorylation of ion-handling proteins and transcriptional regulators that drive excitation–contraction coupling, hypertrophic growth, and arrhythmogenesis. The kinase contains an N‑terminal catalytic domain followed by an autoinhibitory region harboring the CaM-binding element and Thr287 autophosphorylation site, and a C‑terminal association domain that promotes oligomerization into dodecamers and larger assemblies; this architecture supports intersubunit autophosphorylation and permits transition from Ca²⁺‑dependent to autonomous activity through Thr287 phosphorylation, methionine oxidation, and O‑GlcNAcylation, so that CaMKIIδ can remain active after the initiating Ca²⁺ pulse. In ventricular myocytes, CaMKIIδ phosphorylates the L‑type Ca²⁺ channel, ryanodine receptor 2, and phospholamban, which increases sarcolemmal Ca²⁺ influx, enhances sarcoplasmic reticulum Ca²⁺ leak, and accelerates SR refilling, and chronic overactivation of this network leads to diastolic Ca²⁺ overload, delayed afterdepolarizations, and triggered activity that underlie ventricular tachycardia and atrial fibrillation. The same kinase also phosphorylates class IIa histone deacetylase HDAC4, leading to its nuclear export and derepression of MEF2‑dependent gene programs, and thereby links Ca²⁺ and neurohumoral stress to hypertrophic gene expression, fibrosis, and adverse remodeling in pressure overload and neurohormone-driven heart disease. Genetic deletion of CaMKIIδ in mice subjected to transverse aortic constriction preserves cardiac hypertrophy but markedly attenuates the transition to heart failure, reducing chamber dilation, systolic dysfunction, fibrosis, apoptosis, and SR Ca²⁺ leak, whereas transgenic overexpression of CaMKIIδ produces action potential prolongation, spontaneous afterdepolarizations, dilated cardiomyopathy, and increased sudden death, confirming this isoform as a critical transducer of pathological cardiac stress. Human failing and ischemic cardiomyopathy samples show elevated CaMKIIδ expression and autonomous activity and increased phosphorylation of downstream Ca²⁺-handling targets, and pharmacologic CaMKII inhibition in structural heart disease models improves left ventricular function and suppresses arrhythmias. |
| References |
|---|
|
Tech Support
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.