|
Toll Free: (877) 796-6397 -- USA and Canada only -- |
Fax: +1-832-582-8590 Orders: +1-832-582-8158 |
Tech Support: +1-832-582-8158 Ext:3 Please provide your Order Number in the email. |
Technical Data
| Formula | C21H22Cl2FN5O |
|||
| Molecular Weight | 450.34 | CAS No. | 877399-52-5 | |
| Solubility (25°C)* | In vitro | DMSO | 9 mg/mL (19.98 mM) | |
| Water | Insoluble | |||
| Ethanol | Insoluble | |||
|
* <1 mg/ml means slightly soluble or insoluble. * Please note that Selleck tests the solubility of all compounds in-house, and the actual solubility may differ slightly from published values. This is normal and is due to slight batch-to-batch variations. * Room temperature shipping (Stability testing shows this product can be shipped without any cooling measures.) |
||||
Preparing Stock Solutions
Biological Activity
| Description | Crizotinib is a potent inhibitor of c-Met and ALK with IC50 of 11 nM and 24 nM in cell-based assays, respectively. It is also a potent ROS1 inhibitor with Ki value less than 0.025 nM. This compound induces autophagy through inhibition of the STAT3 pathway in multiple lung cancer cell lines. | ||||||
|---|---|---|---|---|---|---|---|
| Targets |
|
||||||
| In vitro | PF-2341066 displays similar potency against c-Met phosphorylation in mIMCD3 mouse or MDCK canine epithelial cells with IC50 of 5 nM and 20 nM, respectivly. This compound shows improved or similar activity against NIH3T3 cells engineered to express c-Met ATP-binding site mutants V1092I or H1094R or the P-loop mutant M1250T with IC50 of 19 nM, 2 nM and 15 nM, respectively, compared with NIH3T3 cells expressing wild-type receptor with IC50 of 13 nM. In contrast, a marked shift in potency of this compound is observed against cells engineered to express c-Met activation loop mutants Y1230C and Y1235D with IC50 of 127 nM and 92 nM, respectively, compared with wild-type receptor. It also potently prevents the phosphorylation of c-Met in NCI-H69 and HOP92 cells, with IC50 of 13 nM and 16 nM, respectively, which express the endogenous c-Met variants R988C and T1010I, respectively. This compound is >1,000-fold selective for the VEGFR2 and PDGFRβ RTKs, >250-fold selective for IRK and Lck, and ∼40- to 60-fold selective for Tie2, TrkA, and TrkB, all compared with c-Met. It is 20- to 30-fold selective for RON and Axl RTKs. In contrast, this compound shows a near-equivalent IC50 of 24 nM against the nucleophosmin (NPM)-anaplastic lymphoma kinase (ALK) oncogenic fusion variant of the ALK RTK expressed by the KARPAS299 human anaplastic large cell lymphoma (ALCL) cell line. It inhibits c-Met–dependent neoplastic phenotypes of cancer cells and angiogenic phenotypes of endothelial cells. This chemical suppresses human GTL-16 gastric carcinoma cell growth with IC50 of 9.7 nM. It induces apoptosis in GTL-16 cells with IC50 of 8.4 nM. It inhibits HGF-stimulated human NCI-H441 lung carcinoma cell migration and invasion with IC50 of 11 nM and 6.1 nM, respectively. It inhibits MDCK cell scattering with IC50 of 16 nM. It prevents HGF-stimulated c-Met phosphorylation, cell survival, and Matrigel invasion with IC50 of 11 nM, 14 nM and 35 nM, respectively. In addition, it prevents serum-stimulated HMVEC branching tubulogenesis (formation of vascular tubes) in fibrin gels. [1] It also potently inhibits NPM-ALK phosphorylation in Karpas299 or SU-DHL-1 ALCL cells with an IC50 of 24 nM. This compound potently prevents cell proliferation, which is associated with G(1)-S-phase cell cycle arrest and induction of apoptosis in ALK-positive ALCL cells with IC50 of 30 nM, but not ALK-negative lymphoma cells. [2] Besides, it prevents osteosarcoma behavior associated with primary tumor growth (i.e., proliferation and survival) as well as metastasis (eg, invasion and clonogenicity). [3] |
||||||
| In vivo | In the GTL-16 model, PF-2341066 reveals the ability to cause marked regression of large established tumors (>600 mm3) in both the 50 mg/kg/day and 75 mg/kg/day treatment cohorts, with a 60% decrease in mean tumor volume over the 43-day administration schedule. In an another study, this compound displays the ability to completely inhibits GTL-16 tumor growth for >3 months, with only 1 of 12 mice exhibiting a significant increase in tumor growth over the 3-month treatment schedule at 50 mg/kg/day. In the NCI-H441 NSCLC model, a 43% decrease in mean tumor volume is observed at 50 mg/kg/day during the 38-day PF-2341066 administration cycle. In the Caki-1 RCC model, a 53% decrease in mean tumor volume is observed to be associated with decreased volume of each tumor by at least 30% at 50 mg/kg/day during the 33-day PF-2341066 administration cycle. This compound also reveals near-complete prevention of the growth of established tumors at 50 mg/kg/day in the U87MG glioblastoma or PC-3 prostate carcinoma xenograft models, with 97% or 84% inhibition on the final study day, respectively. In contrast, this chemical p.o. given at 50 mg/kg/day does not significantly inhibit tumor growth in the MDA-MB-231 breast carcinoma model, or the DLD-1 colon carcinoma model. A significant dose-dependent reduction of CD31–positive endothelial cells is observed at 12.5 mg/kg/day, 25 mg/kg/day, and 50 mg/kg/day in GTL-16 tumors, indicating that inhibition of MVD shows a dose-dependent correlation to antitumor efficacy. This compound displays a significant dose-dependent reduction of human VEGFA and IL-8 plasma levels in both the GTL-16 and U87MG models. Marked inhibition of phosphorylated c-Met, Akt, Erk, PLCλ1, and STAT5 levels is observed in GTL-16 tumors following p.o. administration of this compound.[1] P.o. administration of this chemical to severe combined immunodeficient-Beige mice bearing Karpas299 ALCL tumor xenografts leads to dose-dependent antitumor efficacy with complete regression of all tumors at the 100 mg/kg/d dose within 15 days of initial compound administration. In addition, inhibition of key NPM-ALK signaling mediators, including phospholipase C-gamma, signal transducers and activators of transcription 3, extracellular signal-regulated kinases, and Akt by this compound are observed at concentrations or dose levels, which correlated with inhibition of NPM-ALK phosphorylation and function.[2] This compound prevents osteosarcoma behavior associated with primary tumor growth (eg, proliferation and survival) as well as metastasis (eg, invasion and clonogenicity). In nude mice treated with this chemical via oral gavage, the growth and associated osteolysis and extracortical bone matrix formation of osteosarcoma xenografts are prevented by this compound.[3] Treatment of c-MET-amplified GTL-16 xenografts with 50 mg/kg this compound elicits tumor regression that is associated with a slow reduction in 18F-FDG uptake and decreases expression of the glucose transporter 1, GLUT-1.[4] |
Protocol (from reference)
| Kinase Assay: |
|
|---|---|
| Cell Assay: |
|
| Animal Study: |
|
References
|
Customer Product Validation
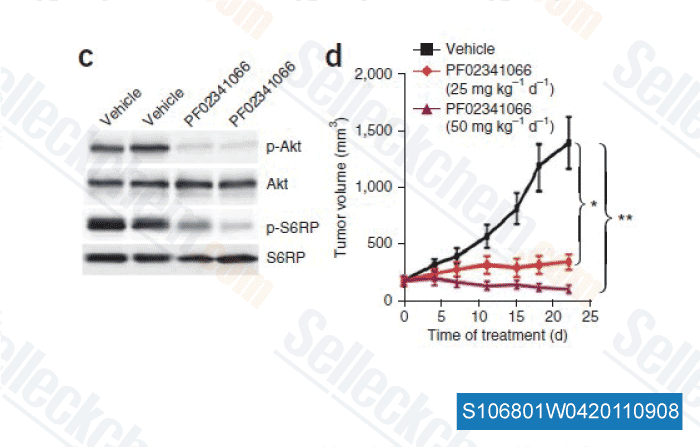
-
Data from [ Nat Med , 2011 , 17, 1116-1120 ]
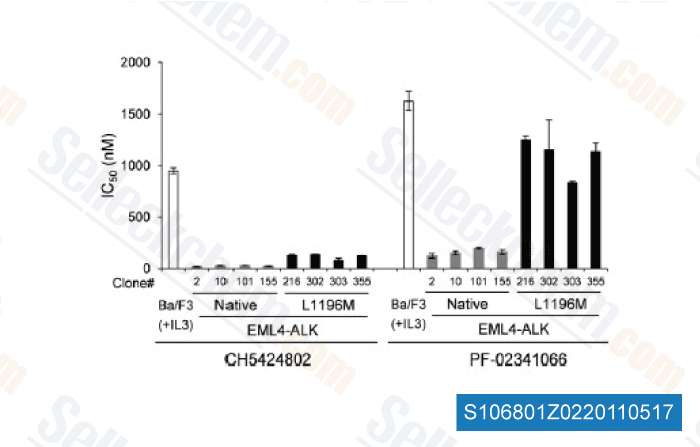
-
Data from [ Cancer Cell , 2011 , 19, 679–690 ]
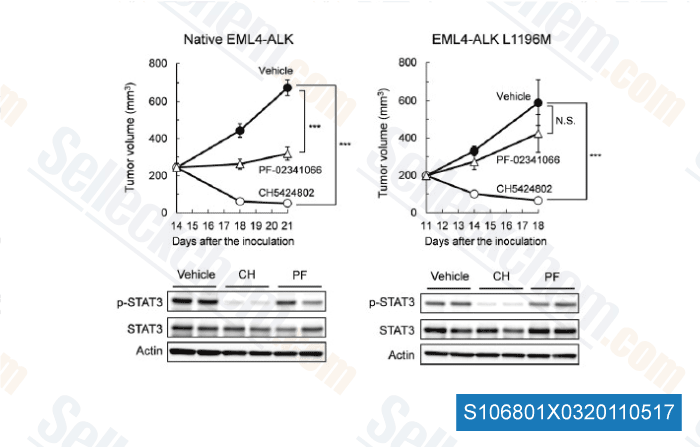
-
Data from [ Cancer Cell , 2011 , 19, 679–690 ]

-
Data from [ J Biomol Screen , 2011 , 16, 141-154 ]
Selleck's Crizotinib Has Been Cited by 534 Publications
| RNase1-driven ALK-activation is an oncogenic driver and therapeutic target in non-small cell lung cancer [ Signal Transduct Target Ther, 2025, 10(1):124] | PubMed: 40246819 |
| A patient-derived T cell lymphoma biorepository uncovers pathogenetic mechanisms and host-related therapeutic vulnerabilities [ Cell Rep Med, 2025, S2666-3791(25)00102-8] | PubMed: 40147445 |
| ARID1A loss enhances sensitivity to c-MET inhibition by dual targeting of GPX4 and iron homeostasis, inducing ferroptosis [ Cell Death Differ, 2025, 10.1038/s41418-025-01510-x] | PubMed: 40369167 |
| Targeting proteostasis in multiple myeloma through inhibition of LTK [ Leukemia, 2025, 10.1038/s41375-025-02682-8] | PubMed: 40634511 |
| Multi-layer stratified oncology platform utilizing transcriptomics, prostate cancer organoids, and modeling of drug response [ J Exp Clin Cancer Res, 2025, 44(1):290] | PubMed: 41094672 |
| Novel selective strategies targeting the BCL-2 family to enhance clinical efficacy in ALK-rearranged non-small cell lung cancer [ Cell Death Dis, 2025, 16(1):194] | PubMed: 40113795 |
| The Src family kinase inhibitor drug Dasatinib and glucocorticoids display synergistic activity against tongue squamous cell carcinoma and reduce MET kinase activity [ Cell Commun Signal, 2025, 23(1):293] | PubMed: 40537792 |
| Synergistic effects of oncogene inhibition and pyruvate dehydrogenase kinase blockade in resistant NSCLC cells [ Biochim Biophys Acta Mol Basis Dis, 2025, 1871(8):168014] | PubMed: 40784600 |
| HGF Overexpression in Mesenchymal Stromal Cell-Based Cell Sheets Enhances Autophagy-Dependent Cytoprotection and Proliferation to Guard the Epicardial Mesothelium [ Int J Mol Sci, 2025, 26(15)7298] | PubMed: 40806435 |
| Therapeutic benefit of the dual ALK/FAK inhibitor ESK440 in ALK-driven neuroblastoma [ Neoplasia, 2025, 60:100964] | PubMed: 39900433 |
RETURN POLICY
Selleck Chemical’s Unconditional Return Policy ensures a smooth online shopping experience for our customers. If you are in any way unsatisfied with your purchase, you may return any item(s) within 7 days of receiving it. In the event of product quality issues, either protocol related or product related problems, you may return any item(s) within 365 days from the original purchase date. Please follow the instructions below when returning products.
SHIPPING AND STORAGE
Selleck products are transported at room temperature. If you receive the product at room temperature, please rest assured, the Selleck Quality Inspection Department has conducted experiments to verify that the normal temperature placement of one month will not affect the biological activity of powder products. After collecting, please store the product according to the requirements described in the datasheet. Most Selleck products are stable under the recommended conditions.
NOT FOR HUMAN, VETERINARY DIAGNOSTIC OR THERAPEUTIC USE.