-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
research use only
VAPB Antibody [L11L13]
Cat.No.: F6454
Application:
Reactivity:
-
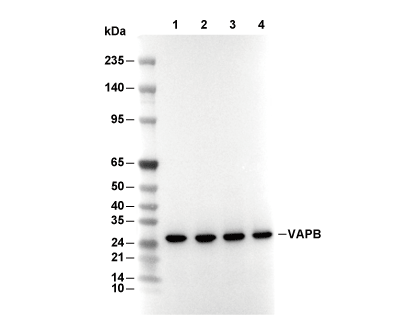 Lane 1: 293T, Lane 2: HepG2, Lane 3: Caco-2, Lane 4: Mouse liver
Lane 1: 293T, Lane 2: HepG2, Lane 3: Caco-2, Lane 4: Mouse liver
Usage Information
| Dilution |
|---|
|
| Application |
|---|
| WB. IP, IHC |
| Reactivity |
|---|
| Human, Mouse |
| Source |
|---|
| Rabbit Monoclonal Antibody |
| Storage Buffer |
|---|
| PBS, pH 7.2+50% Glycerol+0.05% BSA+0.01% NaN3 |
| Storage (from the date of receipt) |
|---|
| -20°C (avoid freeze-thaw cycles), 2 years |
| Predicted MW Observed MW |
|---|
| 27 kDa 27 kDa,15 kDa |
| *Why do the predicted and actual molecular weights differ? The following reasons may explain differences between the predicted and actual protein molecular weight. Post-translational modifications(e.g., phosphorylation, glycosylation); Splice variants and isoforms; Relative charge; Multimerization. |
Biological Description
| Specificity |
|---|
| VAPB Antibody [L11L13] detects endogenous levels of total VAPB protein. |
| Clone |
|---|
| L11L13 |
| Synonym(s) |
|---|
| UNQ484/PRO983, VAPB, Vesicle-associated membrane protein-associated protein B/C, VAMP-B/VAMP-C, VAMP-associated protein B/C, VAP-B/VAP-C |
| Background |
|---|
| VAMP‑associated protein B (VAPB) is an endoplasmic reticulum type IV membrane adaptor of the VAP family that organizes membrane contact sites and couples the ER to diverse organelles and signaling complexes, thereby coordinating lipid transfer, calcium signaling, proteostasis, and neuronal homeostasis. The protein contains an N‑terminal cytosolic MSP (major sperm protein)–like domain that binds FFAT‑motif–containing partners, a central coiled region that supports dimerization and interaction with its paralogue VAPA, and a short C‑terminal transmembrane anchor that embeds VAPB in the ER membrane and positions its interaction platform toward the cytosol. Through its MSP domain, VAPB recruits lipid‑transfer and sorting proteins such as STARD3, OSBP‑related proteins, and WDR44 to the ER surface, promoting formation of ER–endosome, ER–Golgi, and ER–plasma membrane contact sites that facilitate non‑vesicular transfer of cholesterol, phospholipids, and phosphoinositides and support export of newly synthesized cargo from the ER. Interaction with the outer mitochondrial membrane protein PTPIP51 generates a dedicated ER–mitochondria tethering complex in which VAPB–PTPIP51 contacts regulate the proximity of the two organelles, thereby modulating phosphatidylserine and cholesterol exchange, calcium transfer via IP3 receptors and mitochondrial calcium uniporter, and the balance between mitochondrial ATP production, reactive oxygen species levels, and autophagy. VAPB also participates in the unfolded protein response by binding and modulating the ER stress sensor ATF6 and by promoting ERN1/IRE1 activation, linking its scaffold function to transcriptional programs that adjust chaperone expression and ER‑associated degradation capacity under proteotoxic stress. Regulation of phosphoinositide homeostasis and Ca²⁺ signaling by VAPB affects neurite outgrowth and synaptic maintenance, and its broad interaction network at ER contact sites places it as a central coordinator of neuronal membrane dynamics and metabolic signaling. Missense mutation p.P56S in the MSP domain causes a dominantly inherited form of motor neuron disease (ALS8); the mutant protein is aggregation‑prone, unstable, and functionally compromised, and reduction of effective VAPB function (haploinsufficiency) leads to defects in ER–organelle contacts, phosphoinositide balance, Ca²⁺ handling, axonal outgrowth, and ER stress tolerance in motor neurons. Altered VAPB levels and function are also reported in broader ALS cohorts and other neurodegenerative contexts, where disruption of ER–mitochondria tethering and contact‑site signaling contributes to impaired bioenergetics, disturbed calcium homeostasis, and enhanced vulnerability to stress in neurons. |
| References |
|---|
|
Tech Support
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.