-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
research use only
REST Antibody [B2F15]
Cat.No.: F4056
Application:
Reactivity:
-
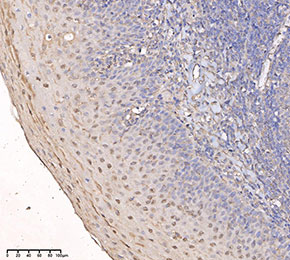 Immunohistochemical analysis of formalin fixed paraffin embedded human tonsils tissue with F4056 at 1:50 dilution.
Immunohistochemical analysis of formalin fixed paraffin embedded human tonsils tissue with F4056 at 1:50 dilution.
Usage Information
| Dilution |
|---|
|
| Application |
|---|
| IHC, IF |
| Reactivity |
|---|
| Human |
| Source |
|---|
| Mouse Monoclonal Antibody |
| Storage Buffer |
|---|
| PBS, pH 7.2+50% Glycerol+0.05% BSA+0.01% NaN3 |
| Storage (from the date of receipt) |
|---|
| -20°C (avoid freeze-thaw cycles), 2 years |
| Predicted MW |
|---|
| 122 kDa |
Biological Description
| Specificity |
|---|
| REST Antibody [B2F15] detects endogenous levels of total REST protein. |
| Clone |
|---|
| B2F15 |
| Synonym(s) |
|---|
| NRSF, XBR, REST, RE1-silencing transcription factor, Neural-restrictive silencer factor, X2 box repressor |
| Background |
|---|
| REST (RE1-silencing transcription factor), also known as NRSF, is a Kruppel-type zinc finger transcriptional repressor that binds the conserved 21–23 bp RE1/NRSE motif and functions as a master negative regulator of neuronal gene expression across development, adult brain plasticity, and neurodegeneration. The protein contains eight Cys2-His2 zinc fingers arranged in a central DNA-binding domain that contacts RE1 elements in promoter and enhancer regions, flanked by N‑ and C‑terminal repression domains that recruit distinct corepressor platforms, including mSin3A–HDAC1/2 and CoREST–HDAC1/2–LSD1–G9a complexes, enabling coordinated histone deacetylation, H3K4 demethylation, and H3K9 trimethylation to compact chromatin and silence target loci. REST occupies thousands of genomic sites in embryonic stem cells, neural progenitors, and differentiated neurons, directly repressing genes encoding ion channels, synaptic vesicle proteins, neurotransmitter receptors, transporters, and neuron-specific microRNAs, and thereby modulates synaptogenesis, axon guidance, membrane excitability, and structural plasticity while also targeting non-neuronal pathways such as JAK–STAT and HIF‑1 signaling in human brain. REST expression is high in pluripotent stem cells and neural progenitor cells, where it maintains a non-neuronal transcriptional program and prevents premature neurogenesis, and declines in a spatially and temporally controlled manner during terminal neuronal differentiation, allowing derepression of neuronal genes and acquisition of mature neuronal phenotypes; persistent REST in progenitors biases fate toward glial lineages and influences gliogenesis. REST abundance and activity are dynamically regulated by casein kinase 1–βTrCP-dependent ubiquitin–proteasome degradation, lysosomal–autophagic turnover, and nucleocytoplasmic trafficking, with context‑dependent nuclear accumulation in mature neurons in response to ischemia, seizures, and Huntington’s disease, or cytoplasmic sequestration by huntingtin–HAP1–RILP complexes under basal conditions. Within aging human cortex and hippocampus, REST reactivates at low levels in excitatory neurons, where it binds promoters of pro‑apoptotic, oxidative stress, and AD‑related genes (including those encoding BAX, PUMA, TNF‑pathway adaptors, γ‑secretase components, and CDK5 regulators) and represses their expression, conferring stress resistance, reduced vulnerability to amyloid and tau pathology, and association with preserved cognition and longevity even in the presence of Alzheimer-type lesions. Loss or reduction of REST in aging neurons correlates with shortened neuronal lifespan and transition from normal cognition to mild cognitive impairment and Alzheimer’s disease, and depletion of REST is also observed in frontotemporal dementia and dementia with Lewy bodies, supporting a general neuroprotective role of REST in late life. In contrast, excessive REST activation in post‑ischemic hippocampal neurons or in temporal lobe epilepsy drives epigenetic silencing of subsets of REST targets such as GluA2, GluN2B, KCC2, and HCN channels, promoting Ca²⁺‑permeable AMPA receptor expression, altered chloride homeostasis, and network hyperexcitability that contribute to excitotoxic neuronal death and epileptogenesis. REST-dependent remodeling also extends to noncoding RNA networks, repressing neuron‑specific microRNAs that themselves target multiple non-neuronal and apoptotic genes, thereby amplifying REST’s reach into metabolic, inflammatory, and survival pathways; transcriptomic analyses in Down syndrome brain, organoids, and neural cells identify REST-target gene sets enriched in JAK–STAT, HIF‑1, axon guidance, and fatty acid metabolism pathways, linking altered REST levels to neurogenic‑to‑gliogenic fate shifts, astrocyte activation, and metabolic imbalance. In neurodegenerative settings, REST integrates hypoxic and inflammatory cues with epigenetic output: in hypoxia and ischemia, nuclear REST is induced and binds RE1 sites at neuronal survival genes, while also engaging with TET3 and NSD3 to promote 5‑hydroxymethylcytosine and H3K36me3 at selected loci, indicating a dual capacity to repress or activate transcription depending on cofactor assembly. |
| References |
|---|
|
Tech Support
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.