
- Bioactive Compounds
- By Signaling Pathways
- PI3K/Akt/mTOR
- Epigenetics
- Methylation
- Immunology & Inflammation
- Protein Tyrosine Kinase
- Angiogenesis
- Apoptosis
- Autophagy
- ER stress & UPR
- JAK/STAT
- MAPK
- Cytoskeletal Signaling
- Cell Cycle
- TGF-beta/Smad
- Compound Libraries
- Antibodies
- Bioreagents
- qPCR
- 2x SYBR Green qPCR Master Mix
- 2x SYBR Green qPCR Master Mix(Low ROX)
- 2x SYBR Green qPCR Master Mix(High ROX)
- Protein Assay
- Protein A/G Magnetic Beads for IP
- Anti-Flag magnetic beads
- Anti-Flag Affinity Gel
- Anti-Myc magnetic beads
- Anti-HA magnetic beads
- Poly FLAG Peptide lyophilized powder
- Protease Inhibitor Cocktail
- Protease Inhibitor Cocktail (EDTA-Free, 100X in DMSO)
- Phosphatase Inhibitor Cocktail (2 Tubes, 100X)
- Cell Biology
- Cell Counting Kit-8 (CCK-8)
- Animal Experiment
- Mouse Direct PCR Kit (For Genotyping)
- New Products
- Contact Us
-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
-
 Other Countries
Other Countries
10058-F4
10058-F4 is a c-Myc inhibitor that specificallly inhibits the c-Myc-Max interaction and prevents transactivation of c-Myc target gene expression. 10058-F4 promotes a caspase-3-dependent apoptosis and modulates autophagy.
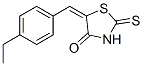
10058-F4 Chemical Structure
CAS: 403811-55-2
Selleck's 10058-F4 has been cited by 95 publications
Purity & Quality Control
Batch:
Purity:
99.95%
99.95
10058-F4 Related Products
| Related Targets | c-Myc | Click to Expand |
|---|---|---|
| Related Compound Libraries | Kinase Inhibitor Library PI3K/Akt Inhibitor Library MAPK Inhibitor Library DNA Damage/DNA Repair compound Library Cell Cycle compound library | Click to Expand |
Signaling Pathway
Choose Selective Myc Inhibitors
Cell Data
| Cell Lines | Assay Type | Concentration | Incubation Time | Formulation | Activity Description | PMID |
|---|---|---|---|---|---|---|
| REH | Function assay | 0-400 µM | 48 h | reduced the metabolic activity, IC50=400 μM | 30957273 | |
| Nalm-6 | Function assay | 0-400 µM | 48 h | reduced the metabolic activity, IC50=430 μM | 30957273 | |
| Jurkat | Function assay | 60 μM | 24 h | c-Myc expression levels treated with VPA (0, 0.8 and 1.6 mM) combined with 60 μM 10058-F4 decreased further compared with the corresponding controls | 25120723 | |
| CCRF-CEM | Function assay | 60 μM | 24 h | c-Myc expression levels treated with VPA (0, 0.8 and 1.6 mM) combined with 60 μM 10058-F4 decreased further compared with the corresponding controls | 25120723 | |
| HL-60 | Function assay | 60 and 100 μM | 24 hours | decreased levels of c-Myc proteins | 17046567 | |
| U937 | Function assay | 60 and 100 μM | 24 hours | decreased levels of c-Myc proteins | 17046567 | |
| NB4 | Function assay | 60 and 100 μM | 24 hours | decreased levels of c-Myc proteins | 17046567 | |
| NB1643 | qHTS assay | qHTS of pediatric cancer cell lines to identify multiple opportunities for drug repurposing: Primary screen for NB1643 cells | 29435139 | |||
| Click to View More Cell Line Experimental Data | ||||||
Biological Activity
| Description | 10058-F4 is a c-Myc inhibitor that specificallly inhibits the c-Myc-Max interaction and prevents transactivation of c-Myc target gene expression. 10058-F4 promotes a caspase-3-dependent apoptosis and modulates autophagy. | |
|---|---|---|
| Targets |
|
| In vitro | ||||
| In vitro | 10058-F4 inhibits growth of leukemic cells and dimerization of Myc and Max. 10058-F4 induces cell-cycle arrest and apoptosis of AML cells. 10058-F4 arrests AML cells at G0/G1 phase, downregulates c-Myc expression and upregulated CDK inhibitors, p21 and p27. Meanwhile, 10058-F4 induces apoptosis through activation of mitochondrial pathway shown by downregulation of Bcl-2, upregulation of Bax, release of cytoplasmic cytochrome C, and cleavage of caspase 3, 7, and 9. Furthermore, 10058-F4 also induces myeloid differentiation, possibly through activation of multiple transcription factors. Similarly, 10058-F4-induced apoptosis and differentiation could also be observed in primary AML cells. [1] 10058-F4 decreases c-Myc protein levels, inhibites proliferation of HepG2 cells likely through upregulation of cyclin-dependent kinase (cdk) inhibitor, p21WAF1 and lowers intracellular levels of [alpha]-fetoprotein (AFP). Treatment with 10058-F4 also downregulates human telomerase reverse transcriptase (hTERT) at the transcriptional level. In addition to inhibiting the proliferation of HepG2 cells, 10058-F4 enhances sensitivity to conventional chemotherapeutic agents, doxorubicin, 5-fluorouracil (5-FU) and cisplatin. [2] | |||
|---|---|---|---|---|
| Cell Research | Cell lines | HL-60, U937, and NB-4 cells | ||
| Concentrations | 0, 30, 60, 90, 120, 150 μM | |||
| Incubation Time | 72 h | |||
| Method | Cells, plated in 96-well plates (105/mL for cell lines and 5 × 105/mL for primary leukemic cells), are treated in triplicate with indicated concentrations of 10058-F4. At various time points, 20 μL 5 mg/mL MTT is added to each well. After incubation at 37°C for 3 hours, the MTT medium is removed and 100 μL DMSO lysis buffer is added. The number of viable cells is assessed by the percentage of absorbance of treated cells relative to that of solvent controls, using 570-nm wavelength on a spectrophotometer. |
|||
| Experimental Result Images | Methods | Biomarkers | Images | PMID |
| Western blot | p-EGFR / HIF-1α / c-Myc / Glut-1 Cyclin D2 / Cyclin D3 / p-21 / c-Myc / p-AKT / AKT PARP / Caspase-3 / Myc |
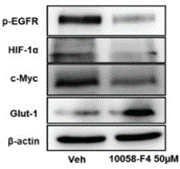
|
30967777 | |
| Growth inhibition assay | Cell viability |
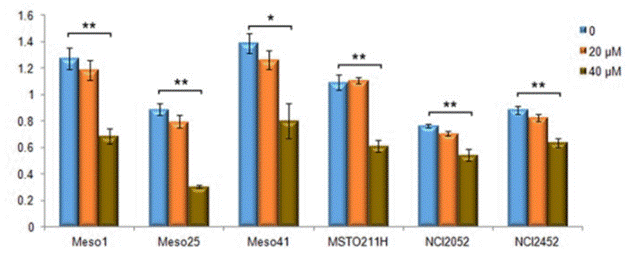
|
28861328 | |
| In Vivo | ||
| In vivo | Peak plasma 10058-F4 concentrations of approximately 300 μM are seen at 5 min and declined to below the detection limit at 360 min following a single iv dose. Plasma concentration versus time data are best approximated by a two-compartment, open, linear model. The highest tissue concentrations of 10058-F4 are found in fat, lung, liver, and kidney. Peak tumor concentrations of 10058-F4 are at least tenfold lower than peak plasma concentrations. Eight metabolites of 10058-F4 are identified in plasma, liver, and kidney. The terminal half-life of 10058-F4 is approximately 1 h, and the volume of distribution is >200 ml/kg. No significant inhibition of tumor growth is seen after i.v. treatment of mice with either 20 or 30 mg/kg 10058-F4.[3] | |
|---|---|---|
| Animal Research | Animal Models | PC-3 and DU145 xenografted SCID mice |
| Dosages | 20 or 30 mg/kg | |
| Administration | i.v. | |
Chemical Information & Solubility
| Molecular Weight | 249.35 | Formula | C12H11NOS2 |
| CAS No. | 403811-55-2 | SDF | Download 10058-F4 SDF |
| Smiles | CCC1=CC=C(C=C1)C=C2C(=O)NC(=S)S2 | ||
| Storage (From the date of receipt) | |||
|
In vitro |
DMSO : 50 mg/mL ( (200.52 mM); Moisture-absorbing DMSO reduces solubility. Please use fresh DMSO.) Ethanol : 1 mg/mL Water : Insoluble |
Molecular Weight Calculator |
|
In vivo Add solvents to the product individually and in order. |
In vivo Formulation Calculator |
||||
Preparing Stock Solutions
Molarity Calculator
In vivo Formulation Calculator (Clear solution)
Step 1: Enter information below (Recommended: An additional animal making an allowance for loss during the experiment)
mg/kg
g
μL
Step 2: Enter the in vivo formulation (This is only the calculator, not formulation. Please contact us first if there is no in vivo formulation at the solubility Section.)
% DMSO
%
% Tween 80
% ddH2O
%DMSO
%
Calculation results:
Working concentration: mg/ml;
Method for preparing DMSO master liquid: mg drug pre-dissolved in μL DMSO ( Master liquid concentration mg/mL, Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug. )
Method for preparing in vivo formulation: Take μL DMSO master liquid, next addμL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O, mix and clarify.
Method for preparing in vivo formulation: Take μL DMSO master liquid, next add μL Corn oil, mix and clarify.
Note: 1. Please make sure the liquid is clear before adding the next solvent.
2. Be sure to add the solvent(s) in order. You must ensure that the solution obtained, in the previous addition, is a clear solution before proceeding to add the next solvent. Physical methods such
as vortex, ultrasound or hot water bath can be used to aid dissolving.
Tech Support
Answers to questions you may have can be found in the inhibitor handling instructions. Topics include how to prepare stock solutions, how to store inhibitors, and issues that need special attention for cell-based assays and animal experiments.
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
* Indicates a Required Field
Tags: buy 10058-F4 | 10058-F4 supplier | purchase 10058-F4 | 10058-F4 cost | 10058-F4 manufacturer | order 10058-F4 | 10058-F4 distributor
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.